Farmacocinética
La medicación se administra para conseguir un efecto terapéutico; para lograrlo, el fármaco debe llegar a las células diana. Esto no resulta difícil para algunos fármacos, como los fármacos tópicos empleados para tratar trastornos cutáneos superficiales. Sin embargo, en otros el proceso de alcanzar las células diana en cantidad suficiente para lograr un cambio fisiológico puede resultar todo un reto, ya que los fármacos se ven expuestos a multitud de barreras y procesos destructivos tras entrar en el organismo. El propósito de este capítulo es analizar los factores que actúan sobre un fármaco durante su intento de alcanzar las células diana.
Conceptos clave
Los conceptos clave proporcionan un breve resumen de los aspectos más importantes de cada uno de los apartados correspondientes dentro del capítulo. Si alguno de estos puntos no está claro, acuda al apartado correspondiente para su revisión.
La farmacocinética se ocupa del desplazamiento del fármaco por el organismo, tras su administración.
Las propiedades fisiológicas de las membranas plasmáticas determinan el desplazamiento de los fármacos por el organismo. Los cuatro procesos de la farmacocinética son la absorción, la distribución, el metabolismo y la eliminación.
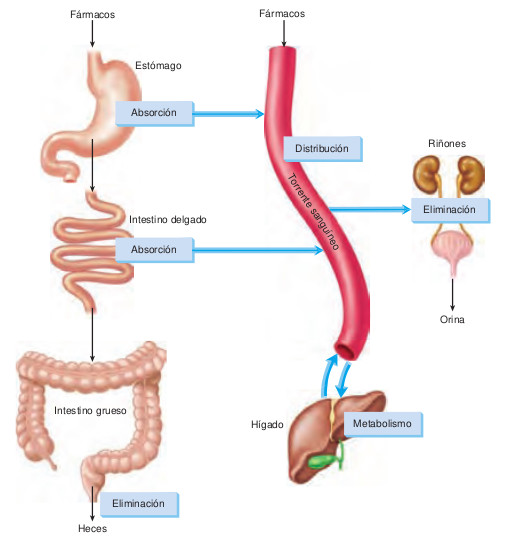
La absorción es el proceso que comprende el desplazamiento de una sustancia desde su punto de administración hasta la circulación. La absorción depende del tamaño de la molécula del fármaco, su liposolubilidad, su grado de ionización y sus interacciones con alimentos u otros medicamentos.
La distribución comprende los métodos empleados para el transporte de los fármacos por el organismo. La distribución depende de la formación de complejos fármaco-proteína y de las barreras especiales, como la placentaria o hematoencefálica.
El metabolismo es el proceso que modifica la actividad del fármaco y facilita su eliminación. Los cambios en el metabolismo hepático pueden influir notablemente en la acción farmacológica.
Los procesos de eliminación expulsan los fármacos del organismo. Los principales órganos excretores son los riñones, pero los fármacos también pueden excretarse por la bilis, los pulmones o las secreciones exocrinas.
La respuesta terapéutica de la mayoría de los fármacos depende de su concentración plasmática. La diferencia entre la concentración mínima eficaz y la concentración tóxica se denomina rango terapéutico.
La semivida plasmática representa la duración de la acción para la mayoría de los fármacos.
La administración de dosis repetidas permite alcanzar una concentración plasmática del fármaco sostenida. Las dosis de carga permiten alcanzar más rápidamente la concentración terapéutica.
Farmacocinética: qué hace el organismo con el fármaco
El término farmacocinética procede de la raíz pharmaco, que significa «medicina», y kinetics, que significa «movimiento». La farmacocinética es, por tanto, el estudio del desplazamiento del fármaco por el organismo. En términos prácticos, describe qué hace el organismo con los medicamentos. La farmacocinética es un pilar de la farmacología y un conocimiento sólido de esta materia permitirá a los profesionales de enfermería comprender mejor y adelantarse a las acciones y los efectos secundarios de los medicamentos administrados a sus pacientes.
En su recorrido hacia las células diana, los fármacos deben enfrentarse a numerosos obstáculos. Para la mayoría de los medicamentos, el principal desafío es atravesar las múltiples membranas que separan el fármaco de las células diana. Por ejemplo, un fármaco tomado por vía oral debe atravesar las membranas plasmáticas de las células de la mucosa digestiva y de las células endoteliales de los capilares para acceder al torrente circulatorio.
Para abandonar la circulación, el fármaco debe atravesar de nuevo las células de los capilares, viajar a través del líquido intersticial y, dependiendo del mecanismo de acción, entrar también a células diana y a orgánulos celulares, como el núcleo, que están rodeados de otras membranas. Estas son sólo algunas de las membranas y barreras que un fármaco debe atravesar antes de desencadenar una respuesta.
Durante el proceso de búsqueda de las células diana y los esfuerzos por atravesar las diversas membranas, los fármacos se ven sometidos a numerosos procesos fisiológicos. Así, en el caso de los fármacos administrados por vía enteral, los ácidos gástricos y las enzimas digestivas degradan a menudo las moléculas del fármaco. Las enzimas presentes en el hígado y en otros órganos pueden provocar cambios químicos en la molécula del fármaco que disminuyan su actividad. Si el organismo considera al fármaco una sustancia extraña, los fagocitos pueden intentar eliminarlo, o puede desencadenarse una respuesta inmunitaria. Los riñones, el intestino grueso y otros órganos también intentan eliminar los medicamentos del organismo.
Estos ejemplos ilustran los procesos farmacocinéticos, es decir, qué hace el organismo con la medicación. Los múltiples procesos farmacocinéticos se clasifican en cuatro categorías: absorción, distribución, metabolismo y eliminación, tal y como ilustra la figura 5.1.
Paso del fármaco a través de las membranas plasmáticas
La variabilidad farmacocinética depende de la capacidad de un fármaco para atravesar las membranas plasmáticas. Con escasas excepciones, los fármacos deben atravesar estas membranas para producir sus efectos y para lograrlo, los fármacos, al igual que otras sustancias químicas, emplean fundamentalmente dos procesos:
- Difusión o transporte pasivo. Desplazamiento de una sustancia química de un área de mayor concentración a una donde la concentración es menor
- Transporte activo. Desplazamiento de una sustancia química en contra de un gradiente de concentración o de un gradiente electroquímico
Las membranas plasmáticas están formadas por una doble capa lipídica en la que se intercalan proteínas y otras moléculas. Esta membrana lipofílica es relativamente impermeable a moléculas de gran tamaño, iones y moléculas polares.
Estas características físicas tienen aplicación directa en la farmacocinética. Así, por lo general, las moléculas de pequeño tamaño, no ionizadas y liposolubles, atravesarán las membranas plasmáticas por difusión simple y alcanzarán las células diana con mayor facilidad. Las sustancias hidrosolubles de bajo peso molecular como la urea, el alcohol y el agua pueden entrar en la célula a través de los poros de la membrana plasmática, mientras que las moléculas de elevado peso molecular, ionizadas e hidrosolubles, tendrán mayor dificultad para atravesar estas membranas. Estas últimas pueden emplear otros métodos para acceder a la célula, como las proteínas transportadoras o el transporte activo. Algunos fármacos no necesitan entrar en la célula para producir sus efectos; se unen a receptores, localizados en la membrana plasmática, lo que activa un segundo mensajero dentro de la célula que es el que produce el cambio fisiológico.
Absorción del fármaco
La absorción es el proceso que comprende el desplazamiento de una sustancia desde su punto de administración hasta la circulación, incluyendo su paso a través de las distintas membranas. Los fármacos pueden absorberse a través de la piel y las mucosas superficiales o pueden atravesar las membranas que recubren los aparatos digestivo o respiratorio. La mayoría de los fármacos, excepto algunas medicaciones tópicas, los antiinfecciosos intestinales y algunos contrastes radiológicos, deben absorberse para producir su efecto.
La absorción es el factor farmacocinético que más influye en el tiempo que necesita un fármaco para producir su efecto. Por norma general, cuanto más rápida es la absorción, más rápido es el inicio de la acción. Así, los fármacos que se emplean en situaciones críticas están diseñados para absorberse en segundos o minutos. En el otro extremo se encuentran otros casos, como la administración intrauterina de la forma de liberación prolongada del anticonceptivo levonorgestrel, administrado mediante un tubo de polietileno que se introduce en el útero. De esta forma, el fármaco se absorbe lentamente y posibilita la protección frente a los embarazos hasta 5 años.
La absorción depende de múltiples factores. El inicio de acción más rápido lo proporcionan los fármacos que se administran por vía intravenosa. Los fármacos que se presentan como jarabe o elixir se absorben más rápido que los comprimidos o las cápsulas. Por lo general, la administración de grandes dosis logra una absorción y un inicio de acción más rápidos que la administración en bajas concentraciones. La motilidad digestiva, la exposición a las enzimas del tubo digestivo y el riego sanguíneo en el punto de administración del fármaco también influyen en la absorción.
La absorción también se ve afectada por el grado de ionización del fármaco, que dependerá del pH del medio en el que se encuentre. El ácido acetilsalicílico supone un excelente ejemplo de los efectos de la ionización en la absorción. En el medio ácido del estómago, el ácido acetilsalicílico se encuentra en su forma no ionizada y, por tanto, resulta fácil su absorción y distribución a través del torrente circulatorio. Sin embargo, a medida que el ácido acetilsalicílico alcanza el medio alcalino del intestino delgado, se ioniza. En esta forma ionizada, este fármaco no puede absorberse ni distribuirse a las células diana tan fácilmente. A diferencia de los fármacos ácidos, los medicamentos que son bases débiles están en su forma no ionizada en un medio alcalino; por tanto, los fármacos básicos se absorben y distribuyen mejor en medios alcalinos como el intestino delgado. Es decir, el pH del medio local influye directamente en la absorción del fármaco al afectar a su ionización. De forma sencilla, puede resultar útil para el estudiante recordar que los ácidos se absorben en ácidos y las bases se absorben en bases.
La absorción de un fármaco también puede verse afectada por sus interacciones con otros fármacos o con alimentos. Se han descubierto múltiples ejemplos de este tipo de interacciones. Así, la administración de tetraciclinas con alimentos o con fármacos que contengan calcio, hierro o magnesio puede retrasar de forma significativa la absorción de este antibiótico. Las comidas ricas en grasa pueden ralentizar la motilidad gástrica de forma notable y, por tanto, retrasar la absorción de los medicamentos orales que se tomen con ellas. Los suplementos dietéticos también pueden afectar a la absorción; los ingredientes habituales de los productos de adelgazamiento fitoterápicos como el aloe vera, la goma guar, la senna y la acedera ejercen un efecto laxante que puede acelerar el tránsito intestinal y reducir así la absorción del fármaco. El profesional de enfermería debe tener en cuenta las interacciones farmacológicas e informar a los pacientes de las combinaciones de medicamentos y alimentos que deben evitar por reducir significativamente la acción del fármaco.
Distribución del fármaco
La distribución supone el transporte de la sustancia farmacológica a través del organismo. El factor básico que determina la distribución es el aporte sanguíneo que reciben los tejidos corporales. El corazón, el hígado, los riñones y el cerebro son los órganos que reciben la mayor cantidad de flujo sanguíneo; la piel, el hueso y el tejido adiposo son los menos vascularizados y, por tanto, resulta más complicado conseguir elevadas concentraciones del fármaco en estas áreas.
Las propiedades físicas del fármaco influyen en gran medida en su desplazamiento a través del organismo. Una característica importante es la liposolubilidad, ya que esta determina la rapidez con que se absorbe un fármaco, interactúa en el torrente sanguíneo, atraviesa las membranas y se asienta en los tejidos corporales. Las barreras que suelen frenar a los fármacos hidrosolubles no afectan a las sustancias liposolubles, por lo que estas llegan mejor a los tejidos corporales.
Algunos tejidos tienen la capacidad de acumular y almacenar los fármacos tras su absorción. La médula ósea, los dientes, los ojos y el tejido adiposo tienen una afinidad, o atracción, especialmente elevada por ciertos medicamentos. Ejemplos de sustancias que se ven atraídas por el tejido adiposo son el tiopental, el diacepam y las vitaminas liposolubles. La tetraciclina se une a las sales de calcio y se acumula en los huesos y los dientes. Una vez almacenados en estos tejidos, los fármacos pueden permanecer en el organismo durante varios meses y liberarse muy lentamente a la circulación.
No todas las moléculas del fármaco que llegan al plasma alcanzan las células diana. Esto se debe a que muchos fármacos establecen una unión reversible con las proteínas plasmáticas, especialmente la albúmina, formando complejos fármacoproteína. Estos complejos son demasiado grandes para atravesar las membranas de los capilares, por lo que el fármaco no puede distribuirse a los tejidos corporales. La fracción del fármaco ligada a proteínas circula por el plasma hasta que se libera o es desplazada de este complejo. Únicamente la fracción libre del fármaco puede alcanzar la célula diana o ser eliminada por los riñones. Algunos fármacos, como el anticoagulante warfarina, tienen un alto grado de unión a las proteínas plasmáticas; el 99% del fármaco localizado en el plasma se presenta en forma de complejos fármaco-proteína y, por tanto, no puede alcanzar las células diana.
Los fármacos y otras sustancias químicas compiten entre ellas por los sitios de unión a las proteínas plasmáticas y la afinidad por estos puntos de unión variará entre las distintas sustancias. Las interacciones de un fármaco con otros fármacos o con alimentos pueden ser consecuencia de que uno de ellos desplace al otro en su unión a las proteínas plasmáticas. En ese caso, el medicamento desplazado alcanza inmediatamente concentraciones elevadas en la sangre y desencadena reacciones adversas. Tomemos como ejemplo el fármaco warfarina; fármacos como el ácido acetilsalicílico o la cimetidina desplazan a este fármaco del complejo fármaco-proteína, lo que elevará las concentraciones plasmáticas de warfarina libre y aumentará drásticamente el riesgo de hemorragia. La mayoría de las guías farmacéuticas informan del porcentaje de medicamento ligado a las proteínas plasmáticas; cuando se administran varios fármacos con un alto grado de unión, el profesional de enfermería debe vigilar atentamente la aparición de reacciones adversas en el paciente.
El cerebro y la placenta disponen de barreras anatómicas especiales que impiden la entrada de múltiples sustancias químicas y medicamentos. Estas barreras se conocen con el nombre de barrera hematoencefálica y barrera placentaria. Algunos medicamentos, como sedantes, ansiolíticos y antiepilépticos, atraviesan fácilmente la barrera hematoencefálica para producir su acción sobre el sistema nervioso central. Por el contrario, la mayoría de los antineoplásicos tiene dificultades para atravesarla, lo que dificulta el tratamiento de los tumores cerebrales.
La barrera placentaria desempeña una importante función protectora, ya que impide el paso de sustancias potencialmente dañinas desde el torrente sanguíneo materno hasta el feto. Sustancias como el alcohol, la cocaína, la cafeína y ciertos medicamentos atraviesan fácilmente la barrera placentaria y pueden causar daños en el feto. En consecuencia, una paciente embarazada no debería tomar ningún tipo de medicamento ni de producto fitoterápico sin consultar antes a su médico. Antes de prescribir un fármaco, el profesional sanitario debería preguntar siempre a las pacientes en edad fértil sobre un posible embarazo.
Metabolismo del fármaco
El metabolismo, también denominado biotransformación, es el proceso de transformación química de un fármaco en una forma que pueda eliminarse del organismo con mayor facilidad.
El metabolismo implica complejas reacciones y rutas bioquímicas que modifican tanto fármacos como nutrientes, vitaminas y minerales. Aunque tiene lugar principalmente en el hígado, los riñones y las células del tracto intestinal también presentan índices metabólicos elevados.
Los medicamentos se ven sometidos a muchos tipos de reacciones bioquímicas en su paso por el hígado; entre ellas, la hidrólisis, la oxidación y la reducción. Durante el metabolismo se añaden cadenas laterales conocidas como conjugados, lo que aumenta la hidrosolubilidad de los fármacos y facilita su expulsión por los riñones.
La mayoría del metabolismo hepático se debe a la acción del sistema enzimático microsomal hepático. Este complejo enzimático se denomina en ocasiones sistema P-450, que recibe su nombre del citocromo P-450, componente clave del sistema.
En cuanto a su papel en la farmacoterapia, las principales acciones de las enzimas microsomales hepáticas son la inactivación del fármaco y la aceleración de su eliminación. Sin embargo, en algunos casos, el metabolismo puede determinar una modificación química que aumente la actividad de la molécula resultante. Por ejemplo, el analgésico opioide codeína que, tras la biotransformación, se convierte en morfina, con mucha mayor capacidad analgésica. De hecho, algunas sustancias, conocidas como profármacos, no tienen actividad farmacológica hasta que el organismo los metaboliza transformándolos en su forma activa; es el caso del benacepril y el losartán.
Cambios en la actividad de las enzimas microsomales hepáticas pueden afectar significativamente al metabolismo del fármaco. Algunos fármacos tienen la capacidad de incrementar la actividad metabólica del hígado, un proceso denominado inducción enzimática. Por ejemplo, el fenobarbital provoca el incremento de la síntesis hepática de enzimas microsomales, de tal manera que aumenta el índice de su propio metabolismo, así como el de otros fármacos metabolizados por el hígado. En estos pacientes, pueden ser necesarias dosis más altas del medicamento para conseguir el efecto terapéutico óptimo.
Algunos pacientes presentan una disminución del metabolismo hepático que puede afectar a la acción del fármaco. Así, la actividad de las enzimas hepáticas suele estar disminuida en niños y ancianos; por tanto, los pacientes pediátricos y geriátricos son más sensibles al tratamiento farmacológico que los pacientes de mediana edad. Los pacientes con un daño hepático importante, por ejemplo, como consecuencia de una cirrosis, precisarán una disminución de la dosis debido a la reducción de la actividad metabólica.
Se han detectado algunos trastornos genéticos en los que los afectados carecen de enzimas metabólicas específicas; en estos pacientes, las dosis de los fármacos deben ajustarse en consecuencia. El profesional de enfermería debe prestar atención a los valores de laboratorio que pueden indicar hepatopatía con objeto de realizar los ajustes necesarios en las dosis.
El metabolismo tiene una serie de consecuencias terapéuticas adicionales. Tras su absorción, los fármacos administrados por vía oral acceden directamente a la circulación portal hepática que transporta la sangre al hígado antes de que se distribuya a otros tejidos corporales. Como consecuencia de este paso de la sangre por la circulación hepática, algunos fármacos pueden metabolizarse completamente e inactivarse antes de alcanzar siquiera la circulación general. Este efecto de primer paso tiene importancia ya que las reacciones metabólicas hepáticas inactivan un gran número de fármacos orales y puede ser necesario considerar vías alternativas de administración que eludan este efecto de primer paso (ej. sublingual, rectal o parenteral).
Eliminación del fármaco
El organismo expulsa los fármacos mediante el proceso de eliminación. La velocidad de eliminación de los medicamentos determina su concentración en el torrente circulatorio y en los tejidos. A su vez, la concentración de los fármacos en el torrente sanguíneo determina la duración de su acción. Algunos estados patológicos, como una hepatopatía o una insuficiencia renal, suelen prolongar la duración de la acción del fármaco en el organismo al interferir con los mecanismos de eliminación naturales; los pacientes con estos trastornos requerirán un ajuste cuidadoso de las dosis.
Aunque la eliminación de los fármacos del organismo puede tener lugar en múltiples órganos y tejidos, el principal órgano excretor es el riñón. Los riñones de un individuo de constitución media filtran diariamente unos 180 L de sangre. La fracción libre de los fármacos, las sustancias hidrosolubles, los electrólitos y las moléculas de bajo peso molecular se filtran fácilmente en el glomérulo. Sin embargo, las proteínas, las células sanguíneas, los conjugados y los complejos fármaco-proteína no pueden filtrarse a causa de su gran tamaño.
Las sustancias químicas y los fármacos filtrados en el corpúsculo renal se ven sometidos al proceso de reabsorción que tiene lugar en el túbulo renal. Los mecanismos de reabsorción son los mismos que los de la absorción en cualquier otro lugar del organismo. Así, los fármacos no ionizados y liposolubles atraviesan fácilmente las membranas de los túbulos renales y vuelven a la circulación, mientras que los fármacos ionizados e hidrosolubles permanecen en el filtrado para su eliminación.
Los complejos fármaco-proteína y las sustancias demasiado grandes para filtrarse en el glomérulo se pueden secretar al túbulo distal de la nefrona. Por ejemplo, sólo el 10% de una dosis de bencilpenicilina se filtra en el glomérulo; el 90% se secreta en el túbulo renal. Al igual que ocurre con la actividad enzimática del metabolismo, la actividad de los mecanismos de secreción es menor en niños y ancianos.
Algunos fármacos pueden eliminarse más rápidamente mediante cambios en el pH del filtrado. Los ácidos débiles como el ácido acetilsalicílico se eliminan más rápidamente cuando el filtrado es ligeramente alcalino, ya que en un medio alcalino este ácido se ioniza, lo que hará que el fármaco permanezca en el filtrado y se elimine por la orina. Las bases débiles como el diacepam se eliminan más rápidamente en un filtrado ligeramente ácido, ya que se ionizan en este medio. Esta relación entre el pH y la eliminación del fármaco puede ser de utilidad en situaciones críticas. Así, se puede administrar bicarbonato sódico para acelerar la eliminación renal de fármacos ácidos, como el ácido acetilsalicílico, en un paciente con sobredosis. El bicarbonato alcalinizará la orina, lo que aumentará la cantidad de ácido acetilsalicílico ionizado, facilitando así su eliminación. Por otra parte, puede estimularse la eliminación del diacepam con la administración de cloruro de amonio, que acidificará el filtrado, aumentando así la eliminación del diacepam.
Una alteración de la función renal puede tener efectos drásticos en la farmacocinética. Los pacientes con insuficiencia renal verán disminuida su capacidad de eliminación de los fármacos, pudiendo retenerlos durante un período de tiempo prolongado. Por tanto, deberán reducirse las dosis administradas a estos pacientes para evitar toxicidades. Cambios pequeños o moderados del estado renal pueden causar un rápido incremento de las concentraciones sanguíneas del fármaco, por lo que el profesional de enfermería debe vigilar constantemente la función renal de los pacientes que reciban fármacos con potencial nefrotóxico o medicamentos con un estrecho margen de seguridad.
Además de los riñones, existen otros importantes órganos excretores. Así, el aparato respiratorio es adecuado para eliminar fármacos que pueden pasar fácilmente al estado gaseoso. La velocidad de eliminación respiratoria depende de los factores que afectan al intercambio gaseoso, como la difusión, la solubilidad del gas y el flujo sanguíneo pulmonar. La eliminación de los anestésicos volátiles tras la cirugía depende fundamentalmente de la actividad respiratoria; a mayor frecuencia respiratoria, mayor eliminación. Por el contrario, la eliminación por esta vía de sustancias hidrosolubles, como el alcohol, depende en mayor medida del aporte sanguíneo a los pulmones; a mayor flujo sanguíneo en los capilares pulmonares, mayor eliminación. Al contrario que otros sistemas de eliminación, los pulmones eliminan la mayoría de los fármacos en su forma original, no metabolizada.
Otro mecanismo de eliminación es la actividad exocrina. Los fármacos hidrosolubles pueden excretarse por la saliva, el sudor o la leche materna. El «gusto raro» que experimentan algunos pacientes tras la administración de fármacos intravenosos es un ejemplo de excreción por la saliva. Otro ejemplo de actividad exocrina es el olor a ajo que puede detectarse cerca de una persona que está sudando y ha comido ajo recientemente.
La eliminación por la leche materna tiene una importancia notable para algunos fármacos esenciales, como la morfina o la codeína, ya que estos pueden alcanzar elevadas concentraciones en la leche materna y afectar al lactante. Las madres que dan el pecho a sus hijos deben consultar siempre con su médico antes de tomar cualquier medicación o suplemento fitoterápico.
Algunos fármacos se eliminan por la bilis, en un proceso conocido como excreción biliar. En muchos casos, los fármacos secretados por la bilis accederán al duodeno y finalmente serán expulsados del organismo por las heces. Sin embargo, la mayor parte de la bilis retorna al hígado mediante la circulación enterohepática, de forma que un porcentaje del fármaco puede recircular varias veces con la bilis. La reabsorción biliar es, en gran medida, responsable de la prolongación de la actividad de los glucósidos cardíacos, algunos antibióticos y las fenotiacinas. Los fármacos que recirculan se metabolizan finalmente en el hígado y se eliminan por los riñones. La circulación y la eliminación de los fármacos mediante la excreción biliar pueden continuar durante varias semanas después de haber finalizado el tratamiento.
Concentración plasmática del fármaco y respuesta terapéutica
La respuesta terapéutica de la mayoría de los fármacos está directamente relacionada con su concentración plasmática.
Aunque conocer la concentración del medicamento en el tejido diana permitiría una predicción más exacta de la acción farmacológica, en la mayoría de los casos esta cantidad es imposible de medir. Por ejemplo, es posible llevar a cabo una prueba de laboratorio para medir la concentración sérica del carbonato de litio mediante la recogida de una muestra de sangre; pero es muy diferente medir la cantidad de este fármaco en las neuronas del SNC. Una práctica de enfermería habitual es la monitorización de las concentraciones plasmáticas de los fármacos con un margen de seguridad estrecho.
La medición de las concentraciones plasmáticas de un fármaco tras la administración de una única dosis permite ilustrar varios de los principios farmacocinéticos más importantes. En ella se pueden apreciar dos concentraciones plasmáticas del fármaco. La primera es la concentración mínima eficaz, la cantidad de fármaco necesaria para obtener un efecto terapéutico. La segunda es la concentración tóxica, la concentración de fármaco que producirá reacciones adversas graves. La concentración plasmática entre la concentración mínima eficaz y la concentración tóxica se denomina rango terapéutico del fármaco. Estos valores tienen gran significancia clínica. Por ejemplo, si el paciente refiere una cefalea intensa y se le administra la mitad de un comprimido de ácido acetilsalicílico, la concentración plasmática permanecerá por debajo de la concentración mínima eficaz y el paciente no experimentará un efecto analgésico; dos o tres comprimidos aumentarán el nivel plasmático del ácido acetilsalicílico hasta el rango terapéutico y el dolor cederá, pero la administración de seis o más comprimidos puede provocar reacciones adversas, como hemorragia digestiva o acúfenos. El objetivo de enfermería será mantener la concentración plasmática de todos los fármacos administrados dentro del rango terapéutico. Aunque en algunos fármacos el rango terapéutico es bastante amplio, en otros la diferencia entre la dosis mínima eficaz y la dosis tóxica puede ser peligrosamente estrecha.
Semivida plasmática y duración de la acción farmacológica
La duración de la acción de un fármaco suele describirse por su semivida plasmática (t½), definida como el período de tiempo necesario para que la concentración plasmática de un medicamento se reduzca a la mitad, tras su administración. Mientras que la semivida de algunos fármacos es, tan sólo, de unos pocos minutos, la de otros es de varias horas o días.
A mayor semivida, más tiempo será necesario para eliminar el medicamento. Por ejemplo, un fármaco con una t½ de 10 horas tardará más en eliminarse, y por tanto su efecto en el organismo será más largo, que el de un fármaco con una t½ de 5 horas.
La semivida plasmática de un fármaco es una variable farmacocinética esencial con importantes aplicaciones clínicas. Así, los fármacos que tienen una semivida relativamente corta, como el ácido acetilsalicílico (t½ = 15-20 minutos), deben administrarse cada 3-4 horas. Los fármacos con una semivida más larga, como el felodipino (t½ = 10 horas), sólo necesitan administrarse una vez al día. En un paciente que padece una hepatopatía o una nefropatía importante, la semivida plasmática de un fármaco aumentará y la concentración del fármaco puede alcanzar niveles tóxicos. En estos pacientes, debe reducirse la frecuencia de administración o la dosis de la medicación.
Dosis de carga y dosis de mantenimiento
Son pocos los fármacos que se administran una única vez. Lo habitual es la administración de dosis repetidas, de forma que el fármaco se va acumulando en el torrente sanguíneo, hasta que finalmente alcanza una meseta, una zona donde su concentración plasmática se mantiene dentro del rango terapéutico de forma continua. Este hecho indica que la cantidad administrada se ha equilibrado con la cantidad de fármaco que se está eliminando y, como consecuencia, los tejidos corporales están recibiendo una concentración terapéutica constante del fármaco. En teoría, son necesarias aproximadamente cuatro semividas para alcanzar este equilibrio. Si el medicamento se administra en perfusión continua, la meseta puede alcanzarse rápidamente y mantenerse con escasas o nulas fluctuaciones de las concentraciones plasmáticas del fármaco.
La meseta puede alcanzarse antes si se administran dosis de carga seguidas de dosis de mantenimiento regulares. Una dosis de carga consiste en una mayor cantidad del fármaco, que suele administrarse sólo una o dos veces, con el fin de alcanzar la concentración plasmática suficiente para inducir rápidamente una respuesta terapéutica. Antes de que las concentraciones plasmáticas puedan caer demasiado, se administran las dosis de mantenimiento intermitentes con el fin de mantenerlas dentro del rango terapéutico. Aunque con esta pauta de administración las concentraciones plasmáticas del fármaco fluctúan, el equilibrio se alcanza casi tan rápidamente como si se administrasen en perfusión continua. Las dosis de carga son especialmente importantes en los fármacos que tienen semividas prolongadas y en aquellas situaciones en las que es crítico alcanzar rápidamente elevadas concentraciones plasmáticas del fármaco, como puede ser el caso de la administración de un antibiótico para tratar una infección grave.