02. Insuficiencia Circulatoria (Shock)
El shock circulatorio puede describirse como un fallo agudo del sistema circulatorio para proporcionar un suministro sanguíneo adecuado a los tejidos periféricos y órganos del cuerpo, lo cual provoca hipoxia celular. Con mayor frecuencia se encuentran hipotensión e hipoperfusión, pero el shock puede ocurrir en presencia de signos vitales normales. El shock no es una enfermedad específica sino un síndrome que puede ocurrir en la evolución de diversas condiciones traumáticas o estados patológicos que ponen en riesgo la vida. Puede ocasionarse por una alteración de la función cardíaca (shock cardiogénico), una disminución del volumen sanguíneo (shock hipovolémico), vasodilatación excesiva y distribución anómala del flujo sanguíneo (shock distributivo), y obstrucción del flujo sanguíneo a través del sistema circulatorio (shock obstructivo). Los tipos principales de shock se resumen en el recuadro 34-1 y se ilustran en la figura 34-8.
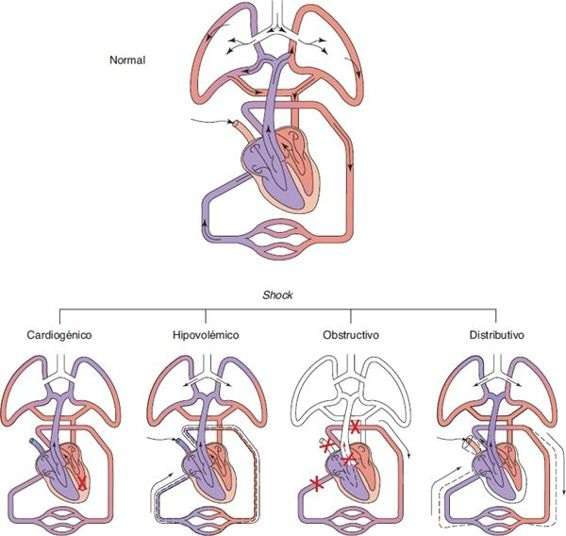
Recuadro 34-1. Clasificación del shock circulatorio
Cardiogénico:
- Lesión miocárdica (infarto de miocardio, contusión).
- Arritmias prolongadas.
- Lesión valvular aguda, defecto del tabique ventricular.
- Cirugía cardíaca.
Hipovolémico:
- Pérdida de sangre total.
- Pérdida de plasma.
- Pérdida de líquido extracelular.
Obstructivo:
- Incapacidad del corazón para llenarse de manera adecuada (taponamiento cardíaco).
- Obstrucción del flujo de salida desde el corazón (embolia pulmonar, mixoma cardíaco, neumotórax o aneurisma disecante).
Distributivo:
- Pérdida del tono vasomotor simpático (shock neurogénico).
- Presencia de sustancias vasodilatadoras en la sangre (shock anafiláctico).
- Presencia de mediadores inflamatorios (shock séptico).
Fisiopatología del shock circulatorio
La insuficiencia circulatoria provoca hipoperfusión de los órganos y tejidos, lo que a su vez ocasiona un suministro insuficiente de oxígeno y nutrientes para la función celular. El cuerpo cuenta con respuestas fisiológicas compensatorias que con el tiempo descompensan en diversos estados de shock si el padecimiento no recibe el tratamiento adecuado en el momento apropiado. Los mecanismos compensatorios más inmediatos son los sistemas simpático y de renina, los cuales están diseñados para mantener el gasto cardíaco y la presión arterial.
Hay 2 tipos de receptores adrenérgicos para el sistema nervioso simpático: α y β. Los β-receptores se subdividen en receptores β1 y β2. La estimulación de los α-receptores ocasiona vasoconstricción; la estimulación de los β1-receptores, un incremento de la frecuencia cardíaca y de la fuerza de contracción miocárdica; y la de los β2-receptores, vasodilatación de los lechos de músculos esqueléticos y relajación de los bronquiolos. En el shock se observa un incremento del flujo simpático que produce un aumento de la liberación de adrenalina y norepinefrina, así como activación de los receptores α y β. De este modo, en casi todos los tipos de shock se encentra un incremento de la frecuencia cardíaca y vasoconstricción. También hay un aumento de la liberación de renina, lo cual causa un incremento de angiotensina II que aumenta la vasoconstricción y provoca el incremento de la retención de agua y sodio mediado por aldosterona en los riñones. Además, hay liberación local de vasoconstrictores, incluidos norepinefrina, angiotensina II, vasopresina y endotelina, todas las cuales contribuyen a la vasoconstricción venosa y arterial.
Los mecanismos compensatorios que recluta el organismo no son efectivos a largo plazo y se tornan perjudiciales cuando el estado de shock es prolongado. La vasoconstricción intensa ocasiona la disminución de la perfusión y un suministro insuficiente de oxígeno para los tejidos. El metabolismo celular se altera, se liberan mediadores inflamatorios vasoactivos como histamina, aumenta la producción de radicales libres de oxígeno y el exceso de iones de hidrógeno y ácido láctico provoca acidez intracelular. Cada uno de estos factores promueve la disfunción o la muerte celular. Si se restablece la función circulatoria, ya sea que el shock sea irreversible o el paciente sobreviva, queda determinado en gran parte a nivel celular.
Finalmente el shock ejerce sus efectos a nivel celular, con fallo de la circulación para proporcionar el oxígeno y los nutrientes necesarios para la producción de ATP en la célula. La célula utiliza ATP para diversos propósitos, incluida la operación de la bomba de sodio-potasio en la membrana que mueve el sodio hacia afuera y el potasio hacia dentro de la célula. Las células utilizan 2 rutas para convertir los nutrientes en energía. La primera es la ruta glucolítica anaeróbica (no depende de oxígeno), la cual se localiza en el citoplasma. La glucólisis convierte la glucosa en ATP y piruvato. La segunda ruta es aeróbica (dependiente de oxígeno), denominada ciclo del ácido cítrico, la cual se localiza en la mitocondria. Cuando se dispone de oxígeno, el piruvato de la ruta glucolítica se mueve hacia la mitocondria y entra al ciclo del ácido cítrico, donde se transforma en ATP y los subproductos metabólicos dióxido de carbono y agua. Cuando se carece de oxígeno, el piruvato no entra al ciclo del ácido cítrico; en cambio, se convierte en ácido láctico. La ruta anaeróbica, aunque permite la producción de energía para continuar en ausencia de oxígeno, es relativamente ineficiente y produce mucho menos ATP que la ruta aeróbica.
En el shock grave, los procesos metabólicos celulares son esencialmente anaeróbicos debido a la poca disponibilidad de oxígeno. Las cantidades excesivas de ácido láctico se acumulan en los compartimentos celular y extracelular, y se producen cantidades limitadas de ATP. Sin la suficiente producción de energía, la función celular normal no puede mantenerse. La bomba de sodio-potasio en la membrana se altera, lo que provoca un exceso de sodio dentro de las células y pérdida intracelular de potasio. El incremento de sodio intracelular ocasiona edema celular y permeabilidad de membrana aumentada. La actividad mitocondrial disminuye de modo importante y las membranas lisosómicas se rompen, con liberación de enzimas que provocan mayor destrucción intracelular. Esto va seguido de la muerte de la célula y liberación de contenido intracelular hacia el espacio extracelular. La destrucción de la membrana de la célula activa la cascada del ácido araquidónico, la liberación de mediadores inflamatorios y la producción de radicales libres de oxígeno, que extienden el daño celular.
La extensión de la lesión microvascular y la disfunción orgánica está determinada en gran parte por la extensión del estado de shock y su duración. Las intervenciones están dirigidas tanto a la prevención como a la intervención temprana, siempre que sea posible.
Shock cardiogénico
El shock cardiogénico ocurre cuando el corazón tiene un fallo al bombear suficiente sangre para satisfacer la demanda corporal (figura 34-8). En lo clínico, se define como la disminución del gasto cardíaco, hipotensión, hipoperfusión e indicaciones de hipoxia de los tejidos, a pesar de un volumen intravascular adecuado. El shock cardiogénico puede presentarse de manera súbita debido a diversas causas, incluidos el infarto de miocardio, la contusión de miocardio, arritmias persistentes y cirugía cardíaca. El shock cardiogénico también puede presentarse como etapa terminal de la cardiopatía coronaria o de la cardiomiopatía.
Fisiopatología
La causa más frecuente del shock cardiogénico es el infarto de miocardio. La mayoría de las personas que fallece debido a shock cardiogénico tiene daño extenso del músculo de contracción del ventrículo izquierdo secundario a un infarto reciente o a una combinación de infartos recientes y antiguos. El shock cardiogénico puede ocurrir con otros tipos de shock debido a flujo sanguíneo inadecuado en las arterias coronarias.
Sin importar la causa, las personas con shock cardiogénico tienen una disminución del volumen latido y del gasto cardíaco, lo cual provoca una perfusión insuficiente para satisfacer las demandas celulares de oxígeno. El gasto cardíaco reducido es consecuencia de una menor contractilidad miocárdica, aumento de la poscarga y precarga excesiva. Los mediadores y neurotransmisores, incluida la norepinefrina, producen un aumento de la resistencia vascular sistémica, lo cual incrementa la poscarga y contribuye al deterioro de la función cardíaca. La precarga, o presión de llenado del corazón, se incrementa conforme la sangre regresa al corazón y se agrega a la sangre que no se bombeó con anterioridad, lo cual causa un aumento del volumen telesistólico del ventrículo izquierdo. La activación del mecanismo de renina-angiotensina-aldosterona empeora tanto la precarga como la poscarga al producir un aumento de la retención de líquido mediada por aldosterona y un incremento de la vasoconstricción mediado por angiotensina II. La mayor resistencia (es decir, la poscarga) a la eyección de sangre desde el ventrículo izquierdo, en combinación con una disminución de la contractilidad miocárdica, provoca el aumento del volumen ventricular telesistólico y de la precarga, lo cual altera aún más la capacidad del corazón para bombear de manera eficaz.
Con el tiempo, la perfusión de las arterias coronarias se altera debido al aumento de la precarga y la poscarga, y la función cardíaca disminuye gracias al poco suministro de oxígeno miocárdico.
Hay un incremento de las presiones intracardíacas debido a la sobrecarga de volumen y a la tensión de la pared ventricular tanto en la diástole como en la sístole. Las presiones excesivas disminuyen la perfusión de las arterias coronarias durante la diástole y el aumento de la tensión de la pared reduce la perfusión de las arterias coronarias durante la diástole. Si el tratamiento no tiene éxito, el shock cardiogénico puede provocar un síndrome de respuesta inflamatoria sistémica. Esto se evidencia por un aumento de la cuenta leucocitaria y de la temperatura, así como por liberación de marcadores inflamatorios como PCR.
Manifestaciones clínicas
Los signos y síntomas del shock cardiogénico incluyen indicaciones de hipoperfusión con hipotensión, aunque el estado previo al shock de hipoperfusión puede ocurrir con una presión arterial normal. Los labios, lechos ungueales y piel se tornan cianóticos debido al estancamiento del flujo sanguíneo y al aumento de la extracción de oxígeno de la hemoglobina mientras pasa a través del lecho capilar. La presión arterial media y la presión arterial sistólica disminuyen debido a un menor volumen latido y hay una presión de pulso estrecha y una presión arterial diastólica casi normal gracias a la vasoconstricción arterial. El gasto urinario disminuye debido a una menor presión de perfusión renal y al aumento de la liberación de aldosterona. La precarga aumentada se refleja en un incremento de PVC y PCPC. Pueden presentarse cambios neurológicos, como alteraciones de la cognición o del estado de alerta, debido a un menor gasto cardíaco y a la poca perfusión cerebral.
Tratamiento
El tratamiento del shock cardiogénico requiere un equilibrio precario y sorprendente entre la mejoría del gasto cardíaco, la disminución de la carga de trabajo y las necesidades de oxígeno del miocardio, además del aumento de la perfusión coronaria. El volumen de líquido debe regularse dentro del intervalo que mantiene la presión de llenado y optimiza el volumen latido en personas que no presentan sobrecarga de líquido. El edema pulmonar y las arritmias deben vigilarse, corregirse y prevenirse para incrementar el volumen latido y disminuir las demandas de oxígeno del corazón. La perfusión de las arterias coronarias se incrementa mediante la promoción de la vasodilatación de dichas arterias, con el aumento de la presión arterial y la disminución de la tensión de la pared ventricular y de las presiones intracardíacas.
El tratamiento farmacológico incluye el empleo de vasodilatadores como nitroprusiato y nitroglicerina. Ambos medicamentos provocan dilatación de las arterias coronarias, lo cual incrementa la entrega miocárdica de oxígeno. Nitroprusiato produce dilatación arterial y venosa, con una disminución del retorno venoso al corazón y una reducción de la resistencia arterial contra la cual el corazón izquierdo debe bombear. En dosis bajas, los efectos principales de nitroglicerina se llevan a cabo en el lecho vascular venoso y en las arterias coronarias. En dosis elevadas, también dilata los lechos arteriales. Ambos medicamentos pueden producir un decremento de la presión arterial diastólica que ocasiona una menor resistencia vascular sistémica (poscarga). La presión arterial sistólica se mantiene a través de un incremento del volumen latido ventricular, el cual se eyecta contra una resistencia vascular sistémica reducida. La mejora de la función cardíaca incrementa el volumen latido y permite la redistribución de la sangre desde el lecho vascular pulmonar hacia la circulación sistémica.
Los fármacos inotrópicos positivos se utilizan para mejorar la contractilidad cardíaca. Tanto la dobutamina como la milrinona son medicamentos eficaces, ya que incrementan la contractilidad y la vasodilatación arterial. La dobutamina es un fármaco sintético que consiste en 2 isómeros, uno de los cuales es un agonista potente de los receptores β1-adrenérgicos y un antagonista de los receptores α1-adrenérgicos, además de ser un agonista débil de los receptores β2-adrenérgicos y de los receptores α1-adrenérgicos. La combinación tiende a producir vasodilatación y una actividad inotrópica positiva. La milrinona incrementa la contractilidad miocárdica mediante el incremento del transporte de Ca++ hacia las células miocárdicas durante un potencial de acción (figura 34-1). El aumento del volumen latido ocasiona una disminución del volumen telesistólico y de la precarga. Con la reducción de las presiones de precarga, mejora la perfusión de las arterias coronarias durante la diástole. De este modo, el volumen latido y el suministro miocárdico de oxígeno mejoran con un incremento mínimo de la demanda de oxígeno por el miocardio. Las catecolaminas aumentan la contractilidad cardíaca, pero deben utilizarse con extrema precaución debido a que además provocan constricción arterial y aumentan la frecuencia cardíaca, lo cual empeora el equilibrio entre el suministro y la demanda miocárdica de oxígeno.
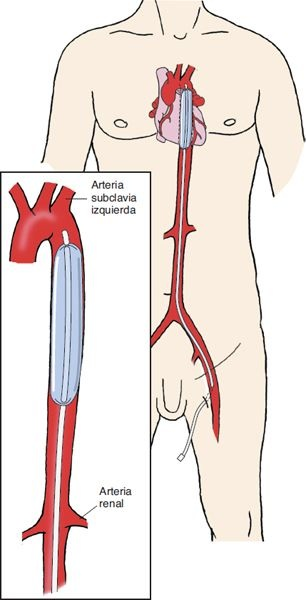
La bomba con balón intraaórtica, también conocida como contrapulsación, refuerza la perfusión coronaria y sistémica, con lo que disminuye la poscarga y la demanda miocárdica de oxígeno. El dispositivo, que bombea en sincronía con el corazón, consiste en un balón de 25,4 cm de largo que se inserta a través de un catéter hacia la aorta descendente (figura 34-9). El balón se programa para inflarse con la diástole ventricular y desinflarse justo antes de la sístole ventricular. La inflación diastólica crea una onda de presión en la aorta ascendente que aumenta el flujo sanguíneo en las arterias coronarias y una onda menos intensa en la región más inferior de la aorta que refuerza la perfusión de los órganos. La desinflación abrupta del balón al inicio de la sístole provoca el desplazamiento del volumen sanguíneo que disminuye la resistencia a la eyección de sangre desde el ventrículo izquierdo. De este modo, aumenta la eficacia de bombeo del corazón y el suministro miocárdico de oxígeno mientras disminuye el consumo de oxígeno por el miocardio.
Cuando el shock cardiogénico se debe a un infarto de miocardio, se utilizan diversas intervenciones agresivas con éxito. Pueden utilizarse tratamiento fibrinolítico, intervención coronaria percutánea o injerto de derivación de arteria coronaria (CABG) para prevenir o tratar el shock cardiogénico. Se espera que la reperfusión de las arterias coronarias mejore la función miocárdica.
Shock hipovolémico
El shock hipovolémico se caracteriza por un volumen sanguíneo disminuido, de tal manera que hay un llenado inadecuado del compartimento vascular (figura 34-8). Se presenta cuando hay una pérdida aguda del 15% al 20% del volumen sanguíneo circulante. La disminución puede ser consecuencia de una pérdida externa de sangre total (ej. hemorragia), plasma (ej. quemadura grave) o líquido extracelular (ej. deshidratación grave o pérdida gastrointestinal de líquido por vómito o diarrea). El shock hipovolémico también puede ser resultado de una hemorragia interna o pérdida en el tercer espacio, en la cual el líquido cambia desde el compartimento vascular al espacio o compartimento intersticial.
Fisiopatología
El shock hipovolémico, que es el tipo más estudiado de shock, con frecuencia se utiliza como prototipo para discusiones sobre las manifestaciones del shock. La figura 34-10 muestra el efecto de retirar sangre del sistema circulatorio durante alrededor de 30 min. Puede retirarse cerca del 10% del volumen total de sangre sin cambiar el gasto cardíaco o la presión arterial. El donador de sangre promedio pierde cerca de 500 ml o el 10% de su sangre sin presentar efectos adversos. Conforme se retiran cantidades crecientes de sangre (10% a 25%), el volumen latido disminuye pero la presión arterial se mantiene gracias al incremento de la frecuencia cardíaca y de la vasoconstricción mediado por el sistema simpático.
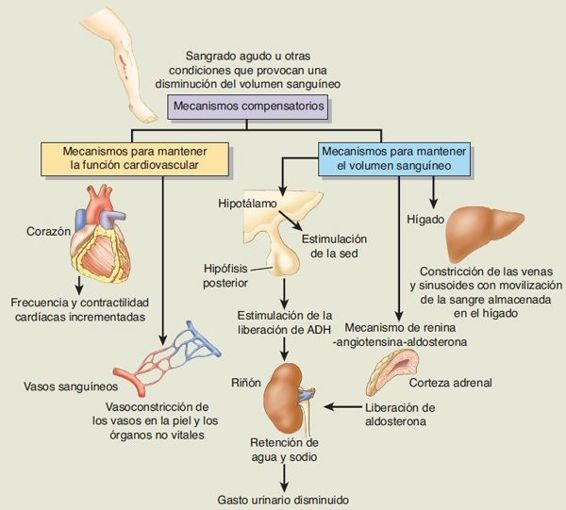
La vasoconstricción provoca una mayor presión diastólica y una presión de pulso estrecha. La presión arterial es el producto del gasto cardíaco y la resistencia vascular sistémica (presión arterial = gasto cardíaco × resistencia vascular sistémica). Un incremento de la resistencia vascular sistémica mantiene la presión arterial media durante un lapso breve a pesar de la disminución del gasto cardíaco. La perfusión de los tejidos y el gasto cardíaco disminuyen antes de que aparezcan los signos de hipotensión. El gasto cardíaco y la presión arterial se reducen a cero cuando alrededor del 30% al 40% del volumen total de sangre se ha retirado.
Mecanismos compensatorios
Sin los mecanismos compensatorios para mantener el gasto cardíaco y la presión arterial, la pérdida de volumen vascular tendría una progresión rápida desde las etapas del shock iniciales hasta las irreversibles. Los mecanismos compensatorios más inmediatos son las respuestas mediadas por el sistema simpático diseñadas para mantener el gasto cardíaco y la presión arterial (figura 34-10). En un lapso de segundos después del inicio de la hemorragia o de la pérdida del volumen sanguíneo, aparecen taquicardia, contractilidad cardíaca incrementada, vasoconstricción y otros signos de actividad simpática y de la médula suprarrenal. La respuesta vasoconstrictora simpática también moviliza sangre que se ha almacenado en el lado venoso de la circulación como un medio para aumentar el retorno venoso hacia el corazón. Se cuenta con una capacidad considerable para almacenar sangre en las grandes venas del abdomen, y cerca de 350 ml de sangre que puede movilizarse en el shock se almacena en el hígado. La estimulación simpática no provoca constricción de los vasos coronarios y cerebrales al inicio, y el flujo sanguíneo hacia el corazón y el cerebro se mantiene en un grado esencialmente normal mientras la presión arterial media permanezca por arriba de 70 mm Hg.
Los mecanismos compensatorios diseñados para restaurar el volumen sanguíneo incluyen absorción de líquido de los espacios intersticiales, conservación de sodio y agua por los riñones y sed. El líquido extracelular se distribuye entre los espacios intersticiales y el compartimento vascular.
Cuando hay una pérdida del volumen vascular, las presiones capilares disminuyen y el agua se retira hacia el compartimento vascular desde los espacios intersticiales. El mantenimiento del volumen vascular se refuerza aún más por mecanismos renales que conservan líquido. La reducción del flujo sanguíneo renal y de la velocidad de filtración glomerular ocasiona la activación del mecanismo de renina-angiotensina-aldosterona, el cual produce un aumento de la reabsorción de sodio por los riñones. La disminución del volumen sanguíneo también estimula los centros del hipotálamo que regulan la liberación de hormona antidiurética (HAD) y la sed. La HAD, también conocida como vasopresina, constriñe las arterias y venas periféricas además de aumentar en gran medida la retención de agua por los riñones. A pesar de que el mecanismo de HAD es más sensible a los cambios en la osmolalidad sérica, una disminución del 10% al 15% del volumen sanguíneo funciona como un estímulo potente para la sed.
Durante las etapas tempranas del shock hipovolémico, la vasoconstricción disminuye el tamaño del compartimento vascular e incrementa la resistencia vascular sistémica. Por lo general, esta respuesta es todo lo que se necesita cuando la lesión es leve y la pérdida de sangre es mínima.
Conforme progresa el shock hipovolémico, la vasoconstricción de los vasos sanguíneos que irrigan la piel, los músculos esqueléticos, riñones y órganos abdominales se torna más intensa, con una disminución adicional del flujo sanguíneo y conversión al metabolismo anaeróbico, que ocasiona lesión celular.
Manifestaciones clínicas
Los signos y síntomas del shock hipovolémico dependen de su gravedad y tienen una relación estrecha con un flujo sanguíneo periférico disminuido y una estimulación simpática excesiva.
Incluyen sed, frecuencia cardíaca aumentada, piel fría y pegajosa, presión arterial arterial reducida, gasto urinario reducido y cambios del estado mental. Los estudios de laboratorio de hemoglobina y hematocrito proporcionan información respecto a la intensidad de la pérdida de sangre y la hemoconcentración debida a deshidratación. El lactato sérico y el pH arterial proporcionan información sobre la gravedad de la acidosis debida al metabolismo anaeróbico. La acidosis metabólica revelada por la medición de los gases en sangre arterial es la prueba diagnóstica estándar de oro. El shock hemorrágico fatal agudo se caracteriza por acidosis metabólica, coagulopatía e hipotermia, seguidas de insuficiencia circulatoria.
El aumento de la frecuencia cardíaca es un signo temprano de shock hipovolémico, ya que el organismo intenta mantener el gasto cardíaco a pesar de la disminución del volumen latido.
Conforme el shock progresa, el pulso se torna débil y filiforme, lo cual indica vasoconstricción y llenado disminuido del compartimento vascular. La sed es un síntoma temprano de shock hipovolémico. A pesar de que la causa subyacente no se comprende del todo, es probable que se relacione con la disminución del volumen sanguíneo y el aumento de la osmolalidad sérica.La presión arterial disminuye en el shock moderado a grave. Sin embargo, hay controversia respecto al valor de las mediciones de la presión arterial en el diagnóstico y manejo tempranos del shock. Esto se debe a que los mecanismos compensatorios tienden a preservar la presión arterial hasta que el shock se encuentra relativamente avanzado. Inclusive, una presión arterial normal no asegura una perfusión y oxigenación adecuadas de los órganos vitales a nivel celular. Esto no implica que la presión arterial no deba vigilarse de modo estrecho en personas en riesgo de desarrollar shock, sino que indica la necesidad de otras medidas de valoración.
Mientras el shock progresa, la respiración se vuelve más rápida y profunda, para compensar la producción incrementada de ácido y la disponibilidad reducida de oxígeno. El volumen intravascular reducido ocasiona un menor retorno venoso al corazón y una PVC disminuida. Cuando el shock se torna grave, las venas periféricas pueden colapsar. La estimulación simpática provoca vasoconstricción intensa de los vasos cutáneos, lo cual se traduce en una piel fría y pegajosa. En el shock hemorrágico, la pérdida de eritrocitos ocasiona palidez de la piel y de las membranas mucosas.
El gasto urinario disminuye con mucha rapidez en el shock hipovolémico. Los mecanismos compensatorios disminuyen el flujo sanguíneo renal como un medio de desviar el flujo de sangre hacia el corazón y el cerebro. La oliguria de 20 ml/h o menos indica perfusión renal inadecuada. La medición continua del gasto urinario es esencial para evaluar el estado circulatorio y de volumen de la persona en shock.
La inquietud, agitación y aprensión son frecuentes en etapas tempranas del shock debido a que el flujo simpático aumenta y a las cifras elevadas de adrenalina. Conforme el shock progresa y el flujo sanguíneo al cerebro disminuye, la inquietud puede reemplazarse por la alteración del estado mental y del despertar. La pérdida del estado de alerta y el coma pueden presentarse si la persona no recibe o responde al tratamiento.
Tratamiento
La duración y cantidad de pérdida de líquido tiene una relación directa con la mortalidad. Por lo tanto, el tratamiento del shock hipovolémico está dirigido a corregir y controlar la causa subyacente, así como a mejorar la perfusión de los tejidos. La pérdida en curso de sangre debe corregirse. Se administra oxígeno para aumentar la entrega de oxígeno a los tejidos. Por lo general, los medicamentos se administran por vía intravenosa. Las mediciones frecuentes del ritmo y de la frecuencia cardíacos, de la presión arterial y del gasto urinario se utilizan para evaluar la gravedad del compromiso circulatorio y para supervisar el tratamiento.
En el shock hipovolémico, el objetivo del tratamiento es restaurar el volumen vascular. Esto puede lograrse mediante la administración intravenosa de líquidos y sangre. Los cristaloides (ej. solución salina isotónica y Ringer lactato) están disponibles con rapidez y son efectivos, por lo menos de manera temporal. Los expansores del volumen plasmático (ej. hidroxietilalmidón [pentastarch] y albúmina coloidal) tienen un alto peso molecular, no necesitan tipificación sanguínea y permanecen en el espacio vascular durante períodos más prolongados que los cristaloides, como la solución salina o de dextrosa. El empleo de cristaloides en lugar de coloides no se ha investigado en estudios clínicos grandes. Por lo tanto, el empleo de uno u otro para disminuir la morbilidad no se ha establecido. La sangre o los productos sanguíneos (paquetes globulares o eritrocitos congelados) se administran con base en los hallazgos hemodinámicos y en el hematocrito. Los líquidos y la sangre se administran mejor con base en los indicadores de volumen como la PVC y el gasto urinario.
Los medicamentos vasoactivos son sustancias capaces de constreñir o dilatar los vasos sanguíneos. Hay una controversia considerable sobre las ventajas o desventajas relacionadas con el consumo de estos fármacos. Como regla general, los medicamentos vasoconstrictores no se utilizan como la primera modalidad terapéutica en el shock hipovolémico y pueden ser nocivos. Estos compuestos se administran sólo cuando el déficit de volumen se ha corregido pero la hipotensión persiste.
Shock distributivo
El shock distributivo o vasodilatador se caracteriza por la pérdida del tono de los vasos sanguíneos, el aumento de tamaño del compartimento vascular y el desplazamiento del volumen vascular lejos del corazón y de la circulación central. En el shock distributivo, la capacidad del compartimento vascular se expande a tal grado que el volumen normal de la sangre no llena el sistema circulatorio (figura 34-8). Por lo tanto, este tipo de shock también se denomina shock normovolémico. La pérdida del tono vascular tiene 2 causas principales: una disminución del control simpático del tono vasomotor o la liberación de sustancias vasodilatadoras en exceso. También ocurre como una complicación de daño vascular resultado de hipotensión prolongada y grave debida a hemorragia, por lo que se conoce como shock hemorrágico en fase tardía o irreversible. Hay 3 estados de shock que comparten el patrón circulatorio básico del shock distributivo: el shock neurogénico, el shock anafiláctico y el shock séptico.
Shock neurogénico
El shock neurogénico es consecuencia de un control simpático disminuido del tono de los vasos sanguíneos debido a un defecto del centro vasomotor en el tallo cerebral o el flujo simpático hacia los vasos sanguíneos. El término shock raquimedular o shock medular describe el shock neurogénico que ocurre en personas con lesión de la médula espinal. El flujo de salida del centro vasomotor puede interrumpirse por alguna lesión cerebral, la actividad depresora de ciertas drogas o fármacos, la anestesia general, hipoxia o ausencia de glucosa (ej. reacción a insulina). El desmayo debido a causas emocionales es una forma transitoria de un flujo simpático alterado. Numerosos fármacos anestésicos generales pueden provocar una reacción parecida a shock neurogénico, en especial durante la inducción, debido a interferencia con la función del sistema nervioso simpático.
La anestesia espinal o la lesión de la médula espinal por arriba de la región torácica media pueden interrumpir la transmisión del flujo de salida desde el centro vasomotor. En contraste con otros estados de shock debidos a la pérdida de volumen de sangre o a una función cardíaca alterada, es común que la frecuencia cardíaca en el shock neurogénico sea más lenta de lo normal, y la piel se encuentra seca y tibia. Este tipo de shock distributivo es raro y, por lo general, es transitorio.
Shock anafiláctico
La anafilaxia es un síndrome clínico que representa la reacción alérgica sistémica más grave. El shock anafiláctico es resultado de una reacción mediada por mecanismos inmunitarios hacia la sangre. Estas sustancias ocasionan vasodilatación de las arteriolas y vénulas además de un incremento marcado de la permeabilidad capilar. Con frecuencia, la respuesta vascular en la anafilaxia se acompaña de edema laríngeo y broncospasmo que ponen en riesgo la vida, colapso circulatorio, contracción del músculo liso uterino y gastrointestinal y urticaria (ronchas) o angioedema.
Etiología
Entre las causas más frecuentes de shock anafiláctico se encuentran las reacciones a medicamentos, como a penicilina; a alimentos, como a nueces y mariscos; y a veneno de insectos. La causa más frecuente es la picadura de insectos del orden Hymenoptera (es decir, abejas, avispas y hormigas de fuego). La alergia al látex ocasiona anafilaxia que pone en riesgo la vida en un segmento creciente de la población. Los trabajadores al cuidado de la salud y otros expuestos al látex han desarrollado sensibilidad a este compuesto, que varía de una urticaria leve, dermatitis por contacto y dificultad respiratoria leve hasta shock anafiláctico. El inicio y la gravedad de la anafilaxia dependen de la sensibilidad de la persona y de la duración y cantidad de la exposición al antígeno.
Manifestaciones clínicas
Los signos y síntomas relacionados con shock anafiláctico inminente incluyen los siguientes:
- Cólicos abdominales.
- Aprensión.
- Sensación de calor o quemazón en la piel.
- Prurito.
- Urticaria (es decir, ampollas o ronchas).
- Tos.
- Asfixia.
- Sibilancias.
- Sensación de opresión torácica.
- Dificultad para respirar.
Después de que la sangre comienza a estancarse en la periferia, hay una disminución precipitada de la presión arterial y el pulso se torna tan débil que es difícil detectarlo. La obstrucción de la vía respiratoria que pone en riesgo la vida puede presentarse como resultado de angioedema laríngeo o espasmo bronquial. Con frecuencia, el shock anafiláctico se desarrolla de modo súbito; la muerte puede ocurrir en un lapso de minutos a menos que se instituya la intervención médica adecuada con prontitud.
Tratamiento
El tratamiento incluye la discontinuación inmediata de la sustancia desencadenante o la institución de medidas para disminuir su absorción (ej. la aplicación de hielo en el sitio de una mordedura/picadura de insecto); la vigilancia estrecha de la función cardiovascular y respiratoria; y el mantenimiento del intercambio de gases respiratorios, el gasto cardíaco y la perfusión de los tejidos. Se administra adrenalina en una reacción anafiláctica debido a que constriñe los vasos sanguíneos y relaja el músculo liso en los bronquiolos, lo cual restaura la función cardíaca y respiratoria. Otras medidas terapéuticas incluyen la administración de oxígeno, fármacos antihistamínicos y corticosteroides. La persona debe situarse en posición supina. Esto tiene importancia extrema, ya que el retorno venoso puede comprometerse de manera grave en la posición sedente. A su vez, esto produce una contracción mecánica del corazón sin pulso y predispone a arritmias. En muchos casos, la muerte ha ocurrido de inmediato después de asumir la posición sedente.
Prevención
La prevención del shock anafiláctico es preferible al tratamiento. Una vez que una persona se ha sensibilizado a algún antígeno, el riesgo de reacciones anafilácticas repetitivas con la exposición subsecuente es alto. Todos los proveedores en atención a la salud deben cuestionar al paciente respecto a sus reacciones medicamentosas previas e informar el nombre del fármaco que recibirá antes de su administración o prescripción. Las personas con hipersensibilidades conocidas deben utilizar algún brazalete o dije de alerta médica y portar una tarjeta de identificación para alertar al personal médico en caso de encontrarse inconscientes o incapaces de proporcionar esta información. Los individuos en riesgo de anafilaxia deben recibir medicamentos de emergencia (ej. auto-inyectores de adrenalina) e instruirse sobre los procedimientos a seguir en caso de que se expongan de manera inadvertida al antígeno desencadenante.
Septicemia y shock séptico
El shock séptico, que es el tipo más frecuente de shock vasodilatador, se relaciona con una infección grave y con la respuesta sistémica a la infección (figura 34-11).
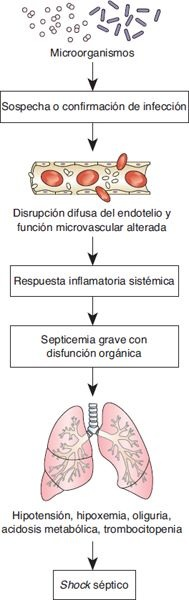
La septicemia se define en la actualidad como la sospecha o confirmación de una infección más un síndrome de respuesta inflamatoria sistémica (ej. fiebre, taquicardia, taquipnea, cuenta leucocitaria elevada, estado mental alterado e hiperglucemia en ausencia de diabetes).
La septicemia grave se define como septicemia con disfunción orgánica (ej. hipotensión, hipoxemia, oliguria, acidosis metabólica, trombocitopenia u obnubilación).
El shock séptico se define como septicemia grave con hipotensión, a pesar de la reanimación con líquidos.
Se estima que la septicemia ocurre en 500 personas cada día en Estados Unidos. La incidencia creciente se ha atribuido a un mejor conciencia sobre el diagnóstico, al incremento de la cantidad de microorganismos resistentes, al número creciente de personas inmunocomprometidas y adultos de edad avanzada, y al mayor empleo de procedimientos invasivos. Con la intervención temprana y los avances en los métodos terapéuticos, la tasa de mortalidad ha disminuido. No obstante, la cantidad de fallecimientos ha aumentado debido a una mayor prevalencia.
Fisiopatología
La patogénesis de la septicemia implica un proceso complejo de activación celular que ocasiona la liberación de mediadores proinflamatorios, como citocinas; el reclutamiento de neutrófilos y monocitos; la implicación de reflejos neuroendocrinos; y la activación de los sistemas fibrinolíticos, del complemento y de la coagulación. El inicio de la respuesta comienza con la activación del sistema inmune innato por receptores de reconocimiento de patrón (ej. receptores tipo Toll [RTT]) que interactúan con moléculas específicas presentes en los microorganismos. La unión de los RTT a los epítopes en los microorganismos estimula la transcripción y liberación de diversos mediadores proinflamatorios y antiinflamatorios. Dos de estos mediadores, FNT-α e interleucina-1, están implicados en la adhesión leucocitaria, la inflamación local, la activación de neutrófilos, la supresión de la eritropoyesis, la generación de fiebre, taquicardia, acidosis láctica, anomalías de ventilación-perfusión y otros signos de septicemia que ya se señalaron con anterioridad. A pesar de que los neutrófilos eliminan microorganismos, también lesionan el endotelio mediante la liberación de mediadores que incrementan la permeabilidad vascular. Además, las células endoteliales activadas liberan óxido nítrico, un vasodilatador potente que actúa como mediador clave en el shock séptico.
Otro aspecto importante de la septicemia es una alteración del equilibrio entre procoagulación-anticoagulación con un incremento de los factores procoagulantes y una disminución de los factores anticoagulantes. El lipopolisacárido en la superficie de los microorganismos estimula las células endoteliales que recubren por dentro a los vasos sanguíneos para aumentar la producción del factor tisular, lo cual activa la coagulación. Entonces, el fibrinógeno se convierte en fibrina y promueve la formación de trombos microvasculares que amplifican la lesión de los tejidos. Además, la septicemia disminuye las concentraciones de proteína C, proteína S, antitrombina III e inhibidor de la vía del factor tisular, sustancias que modulan e inhiben la coagulación. El lipopolisacárido y FNT-α también disminuyen la síntesis de trombomodulina y receptor endotelial de proteína C, lo que altera la activación de proteína C y aumenta la síntesis de inhibidor del activador de plasminógeno 1, que ocasiona la alteración de la fibrinólisis.
Manifestaciones clínicas
De manera típica, la septicemia y el shock séptico se manifiestan como hipotensión y piel caliente y ruborizada. Mientras que otras formas de shock (es decir, cardiogénico, hipovolémico y obstructivo) se caracterizan por un incremento compensatorio de la resistencia vascular sistémica, con frecuencia, el shock séptico se presenta con una disminución de la resistencia vascular sistémica. Hay hipovolemia secundaria a dilatación arterial y venosa, además de fuga de plasma hacia los espacios intersticiales. Los cambios abruptos de la cognición y el comportamiento se deben a un flujo sanguíneo cerebral disminuido y pueden ser indicaciones tempranas de shock séptico. Sin importar la causa subyacente, se encuentran fiebre y cuenta leucocitaria elevada. Un cifra alta de lactato en suero o acidosis metabólica indican metabolismo anaeróbico debido a hipoxia de los tejidos o a disfunción celular y metabolismo alterado en las células. La hipoxia de los tejidos ocasiona la producción y activación continuas de mediadores inflamatorios, lo cual provoca un incremento adicional de la permeabilidad vascular, regulación vascular y hemostasia alteradas.
Tratamiento
El tratamiento de la septicemia y del shock séptico está enfocado en el control de la causa y el soporte de la circulación. El consumo temprano de antibióticos es esencial, seguido de tratamiento antibiótico específico para el microorganismo infeccioso. Sin embargo, los antibióticos no tratan la respuesta inflamatoria de la infección. Por ello, el estado cardiovascular de la persona debe recibir soporte para incrementar la entrega de oxígeno a las células y prevenir el avance de la lesión celular. La administración rápida y agresiva de líquido es necesaria para compensar la formación del tercer espacio. De igual manera, el empleo agresivo de fármacos vasoconstrictores, como vasopresina, norepinefrina y fenilefrina, es necesario para contrarrestar la vasodilatación provocada por los mediadores inflamatorios. Un inotrópico positivo, como la dobutamina o la milrinona, puede utilizarse para aumentar el gasto cardíaco. La valoración continua de oxígeno, PVC, saturación de oxígeno venosa mixta o central, presión arterial media y gasto urinario, así como las mediciones por laboratorio de hemocultivos, lactato sérico, déficit de base y pH se utilizan para evaluar la progresión de la septicemia y la adecuación del tratamiento.
Entre los avances más recientes en el tratamiento de la septicemia se encuentran el empleo de tratamiento intensivo con insulina para la hiperglucemia y la administración de proteína C activada recombinante humana. Se ha demostrado que el tratamiento intensivo con insulina que mantuvo las concentraciones de glucosa en sangre entre 80 mg/dl y 110 mg/dl (4,4 mmol/l a 6,1 mmol/l) provocó una menor mortalidad y morbilidad en comparación con la terapia convencional que mantuvo las cifras sanguíneas de glucosa entre 180 mg/dl y 200 mg/dl (10 mmol/l a 11 mmol/l). La hiperglucemia tiene potencial lesivo debido a que actúa como procoagulante, induce apoptosis, altera la función de los neutrófilos, incrementa el riesgo de infecciones y afecta la recuperación de las heridas. La proteína C activada recombinante humana (drotrecogina α), un factor anticoagulante natural que actúa mediante inactivación de los factores de coagulación Va y VIII, es el primer compuesto que ha demostrado ser eficaz en el tratamiento de la septicemia. Además de su actividad anticoagulante, la proteína C activada tiene propiedades antiinflamatorias, incluido el bloqueo de la producción de citocinas por los monocitos y el bloqueo de la adhesión celular. La proteína C activada también tiene actividad antiapoptótica que puede contribuir a su efectividad. El empleo de corticosteroides, otrora considerado la base del tratamiento de la septicemia, aún es controvertido. Se cuenta con poca o ninguna evidencia de que el empleo de corticosteroides pueda mejorar la evolución del paciente. Sólo debe considerarse cuando la reanimación con líquidos y el soporte vasoactivo no han demostrado mejorar el estado de la persona con septicemia.
Shock obstructivo
El término shock obstructivo describe al shock circulatorio resultante de la obstrucción mecánica del flujo de sangre a través de la circulación central (grandes venas, corazón o pulmones; figura 34-8).
El shock obstructivo puede ocasionarse por diversas condiciones, como un aneurisma aórtico disecante, taponamiento cardíaco, neumotórax, mixoma auricular y evisceración del contenido abdominal hacia la cavidad torácica debido a un hemidiafragma roto. La causa más frecuente de shock obstructivo es la embolia pulmonar.
El resultado fisiológico primordial del shock obstructivo es un incremento de la presión del corazón derecho secundario a función ventricular derecha alterada. Las presiones se encuentran elevadas a pesar de un retorno venoso alterado hacia el corazón. Se encuentran signos de insuficiencia cardíaca derecha, como elevación de PVC y distensión de las venas yugulares. Las modalidades terapéuticas se enfocan en la corrección de la causa del padecimiento, con frecuencia mediante intervenciones quirúgicas como embolectomía pulmonar, pericardiocentesis (es decir, la eliminación de líquido del saco pericárdico) para taponamiento cardíaco o la inserción de un tubo torácico para corrección de un neumotórax a tensión o un hemotórax. En la embolia pulmonar grave o masiva, pueden utilizarse fármacos fibrinolíticos para degradar los coágulos que provocan la obstrucción.
Complicaciones del shock
El shock destruye diversos sistemas corporales. Las 5 complicaciones principales del shock grave son las siguientes:
- Lesión pulmonar.
- Insuficiencia renal aguda.
- Ulceración gastrointestinal.
- Coagulación intravascular diseminada (CID).
- Síndrome de disfunción orgánica múltiple (SDOM).
Estas complicaciones del shock son graves y, con frecuencia, fatales.
Lesión pulmonar aguda/síndrome de dificultad respiratoria aguda
La lesión pulmonar aguda/síndrome de dificultad respiratoria aguda (LPA/SDRA) es una forma potencialmente fatal de lesión pulmonar que puede ser causa o efecto del shock. El SDRA es un aspecto más grave de la LPA y se diferencia de manera primordial para propósitos de intervención temprana, prevención e investigación.
LPA/SDRA está marcada por el inicio rápido de disnea profunda que ocurre de modo usual 12 h a 48 h después del accidente inicial. La frecuencia y el esfuerzo respiratorios aumentan. El análisis de los gases en sangre arterial establece la presencia de hipoxemia profunda que es refractaria a la suplementación con oxígeno. La hipoxemia se debe a la alteración del equilibrio entre la ventilación y la perfusión, así como a una difusión muy reducida de los gases sanguíneos a través de las membranas alveolares engrosadas.
La causa exacta de LPA/SDRA se desconoce. Se piensa que los neutrófilos tienen un papel central en su patogénesis. Se considera que la activación y acumulación de neutrófilos mediada por citocinas en la vasculatura pulmonar y la lesión endotelial subsecuente ocasionan la fuga de líquido y de proteínas plasmáticas hacia el intersticio y los espacios alveolares. La fuga de líquido produce atelectasias, altera el intercambio de gases y causa rigidez pulmonar, por lo que son más difíciles de inflar. Las anomalías de la producción, composición y función del surfactante pueden contribuir al colapso alveolar y las anomalías del intercambio de gases. La vasodilatación inadecuada y la vasoconstricción empeoran la incompatibilidad entre ventilación y perfusión.
Las intervenciones para LPA/SDRA se enfocan en el incremento de la concentración de oxígeno en el aire inspirado y el soporte de la ventilación a través de medios mecánicos para optimizar el intercambio de gases mientras se evita la toxicidad por oxígeno y se previene un mayor daño pulmonar. A pesar de que la administración de concentraciones elevadas de oxígeno mediante soporte ventilatorio mecánico de presión alta y presión positiva al final de la espiración pueden corregir la hipoxemia, la tasa de mortalidad varía del 35% al 40%. Las causas principales son el incidente desencadenante y el fallo orgánico múltiple.
Insuficiencia renal aguda
Los túbulos renales poseen una vulnerabilidad particular a la isquemia y la insuficiencia renal aguda es un factor importante en la mortalidad debida a shock grave. La mayoría de los casos de insuficiencia renal aguda son secundarios a una perfusión renal alterada o a lesión directa de los riñones. El grado de daño renal se relaciona con la gravedad y duración del shock. En condiciones normales, el riñón es capaz de tolerar la isquemia intensa durante 15 min a 20 min. Con mayor frecuencia, la disfunción renal se observada después de shock grave es la necrosis tubular aguda. Porlo general, la necrosis tubular aguda es reversible, aunque el retorno a una función renal normal puede requerir semanas o meses. La vigilancia continua del gasto urinario durante el shock proporciona un medio para evaluar el flujo sanguíneo renal. La vigilancia frecuente de las concentraciones séricas de creatinina y de nitrógeno ureico también proporcionan información valiosa respecto al estado renal.
Los mediadores implicados en el shock séptico son vasoconstrictores poderosos capaces de activar el sistema nervioso simpático y provocar coagulación intravascular. Se ha demostrado que desencadenan todos los mecanismos fisiológicos por separado que contribuyen al inicio de la insuficiencia renal aguda.
Complicaciones gastrointestinales
El tracto gastrointestinal también es vulnerable a la isquemia debido a los cambios en la distribución del flujo sanguíneo hacia su superficie mucosa. En el shock, hay una constricción diseminada de los vasos sanguíneos que irrigan el tracto gastrointestinal, lo cual provoca una redistribución del flujo sanguíneo y una disminución grave de la perfusión de la mucosa. Las personas pueden presentar pérdida del apetito, náuseas o vómito. Las lesiones superficiales de la mucosa del estómago y el duodeno pueden desarrollarse en un lapso de horas a partir de un traumatismo grave, septicemia o quemaduras. Puede haber obstrucción intestinal o sangrado después de la disminución de la perfusión en el shock. Por lo general, la hemorragia tiene su inicio entre los 2 y 10 días posteriores al episodio original y con frecuencia comienza sin advertencia. La poca perfusión del tracto gastrointestinal se agrava por la entrada de bacterias intestinales hacia el torrente sanguíneo, lo cual contribuye al desarrollo de septicemia y shock.
Los antagonistas del receptor tipo 2 de histamina, los inhibidores de la bomba de protones o el sucralfato pueden administrarse de manera profiláctica para prevenir la ulceración gastrointestinal causada por el choque. Las sondas nasogástricas, cuando se aúnan a la succión intermitente, también ayudan a disminuir la acumulación de iones de hidrógeno en el estómago.
Coagulación intravascular diseminada
La coagulación intravascular diseminada (CID) se caracteriza por la activación extendida del sistema de coagulación con la formación resultante de coágulos de fibrina y oclusión trombótica de los vasos de pequeño y mediano calibre. La formación sistémica de fibrina provoca un incremento de la generación de trombina, la supresión simultánea de los mecanismos anticoagulantes fisiológicos y la remoción retrasada de fibrina como consecuencia de una fibrinólisis alterada. En lo clínico se informa que la CID manifiesta se presenta en hasta 1 de cada 1.000 personas en Estados Unidos. Con en otras respuestas inflamatorias sistémicas, se piensa que el desequilibrio entre la coagulación y la fibrinólisis está mediado por mediadores inflamatorios y citocinas.
La contribución de CID a la morbilidad y mortalidad en la septicemia depende de la condición clínica subyacente y la intensidad de la alteración de la coagulación. La disminución de las plaquetas y los factores de coagulación incrementan el riesgo de sangrado. El depósito de fibrina en la vasculatura de los órganos contribuye al daño isquémico y el fallo orgánico. No obstante, aún es incierto si la CID es un factor predictivo de una evolución desfavorable o simplemente un marcador de la gravedad de la condición subyacente causante de CID.
El manejo de la CID inducida por septicemia se enfoca en el tratamiento del padecimiento subyacente y en las medidas para interrumpir el proceso de coagulación. Puede utilizarse el tratamiento anticoagulante, así como la administración de productos hemáticos.
Síndrome de disfunción orgánica múltiple
El síndrome de disfunción orgánica múltiple (SDOM) representa la presencia de una función orgánica alterada en un paciente con enfermedad aguda, de tal manera que la homeostasis no puede mantenerse sin intervención. Como lo implica su nombre, es común que el SDOM afecte múltiples sistemas orgánicos, incluidos los riñones, pulmones, hígado, cerebro y corazón. El SDOM es una complicación que pone en riesgo la vida, en particular en el shock, de modo especial el shock séptico.
Se ha informado que es la causa más frecuente de muerte en la unidad de cuidados intensivos. Las tasas de mortalidad varían entre el 30% y el 100%, según la cantidad de órganos implicados. Las tasas de mortalidad se incrementan con el número creciente de órganos que fallan. Una tasa de mortalidad elevada se relaciona con fallo del cerebro, el hígado, los riñones y los pulmones. La patogénesis del SDOM no se ha comprendido por completo y, por lo tanto, el manejo actual es principalmente de soporte. Los factores de riesgo principales para el desarrollo del SDOM son el traumatismo grave, la septicemia, períodos prolongados de hipotensión, disfunción hepática, intestino infartado, edad avanzada y abuso de alcohol. Las intervenciones para fallo orgánico múltiple se enfocan en el soporte de los órganos afectados.