04. Trastornos de la función de la corteza suprarrenal
Control de la función de la corteza suprarrenal
Las glándulas suprarrenales son estructuras bilaterales pequeñas que pesan alrededor de 5 g cada una y se ubican en el retroperitoneo, sobre el polo superior de cada riñón (figura 49-9).
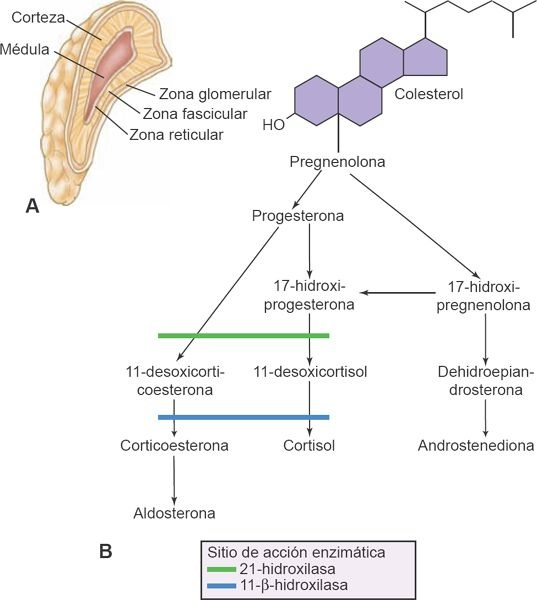
La médula o porción interna de la glándula (que constituye alrededor del 20% de cada glándula suprarrenal) secreta adrenalina y noradrenalina, y forma parte del sistema nervioso simpático. La corteza constituye la mayor parte de la glándula suprarrenal (alrededor del 80%), y es responsable de secretar 3 tipos de hormonas: los glucocorticoides, los mineralocorticoides y los andrógenos suprarrenales. Puesto que el sistema nervioso simpático también secreta adrenalina y noradrenalina, la función de la médula suprarrenal no es esencial para la vida, pero sí lo es la función de la corteza suprarrenal. La pérdida total de la función cortical suprarrenal resulta mortal en pocos días o semanas si no recibe tratamiento. Esta sección del capítulo describe la síntesis y la función de las hormonas de la corteza suprarrenal, y los efectos de la insuficiencia y la función exagerada de la corteza suprarrenal.
Biosíntesis, transporte y metabolismo
En la corteza suprarrenal se producen más de 30 hormonas. Entre éstas, la aldosterona es el mineralocorticoide principal el cortisol (hidrocortisona) es un glucocorticoide principal y los andrógenos son las hormonas sexuales principales. Todas las hormonas de la corteza suprarrenal tienen una estructura semejante, en el sentido de que son esteroides y se sintetizan a partir del acetato y el colesterol. Cada uno de los pasos implicados en la síntesis de las distintas hormonas requiere una enzima específica (figura 49-9). La HACT que secreta el lóbulo anterior de la hipófisis controla la secreción de los glucocorticoides y de los andrógenos suprarrenales.
El cortisol, la aldosterona y los andrógenos suprarrenales se secretan en estado libre y se unen a las proteínas del plasma para ser transportados por el sistema circulatorio. El cortisol se une en mayor medida a la globulina de unión a los corticoesteroides y en menor grado a la albúmina. La aldosterona y los andrógenos circulan en su mayoría unidos a la albúmina. Se ha sugerido que la reserva de hormonas unidas a proteínas pudiera prolongar la duración de su acción al retrasar su eliminación metabólica.
El sitio principal para el metabolismo de las hormonas de la corteza suprarrenal es el hígado, órgano en que sufren distintas conversiones metabólicas antes de conjugarse y volverse hidrosolubles. Se eliminan después, ya sea en la orina o la bilis.
Andrógenos suprarrenales
Los andrógenos suprarrenales se sintetizan de manera principal en la zona reticular y en la zona fascicular de la corteza (figura 49-9A). Estas hormonas sexuales quizá ejercen poco efecto sobre la función sexual normal. A pesar de esto, existe evidencia de que los andrógenos suprarrenales (los más importantes son la dehidroepiandrosterona [DHEA] y su sulfato [DHEAS]) contribuyen al crecimiento juvenil del vello corporal, en particular el vello púbico y axilar en la mujer. También pudieran desempeñar algún papel en la economía de las hormonas esteroides en la mujer embarazada y la unidad fetoplacentaria. El DHEAS se está utilizando cada vez más para el manejo tanto de la enfermedad de Addison como en adultos con disminución de las concentraciones de esa misma sustancia. Los andrógenos suprarrenales tienen relevancia fisiológica en la mujer con enfermedad de Addison y debe considerarse su restitución con 25 mg a 50 mg de DHEAS por día. Puesto que los testículos sintetizan estas hormonas, no hay razón para utilizarlas en el varón. Las concentraciones de DHEAS declinan hasta alrededor del 10% al 20% de las concentraciones de un individuo de 20 años al alcanzar los 80 años de edad (adrenopausia). El valor de la restitución rutinaria del DHEAS en la adrenopausia en gran medida no está probado. Las concentraciones de DHEAS pueden representar otro marcador del envejecimiento, puesto que participa en los sistemas cardiovascular, inmunitario y endocrino, y puede ser un indicador de tendencia para la prevención de problemas específicos del envejecimiento.
Mineralocorticoides
Los mineralocorticoides desempeñan un papel esencial en la regulación de las concentraciones del potasio y el sodio, y en el equilibrio del agua. Se sintetizan en la zona glomerular, la capa externa de células de la corteza suprarrenal. La secreción de aldosterona se encuentra regulada por el mecanismo reninaangiotensina y por las concentraciones del potasio en sangre. El aumento de las concentraciones de la aldosterona promueve la retención de sodio en los túbulos distales del riñón, al tiempo que aumenta las pérdidas urinarias de potasio.La aldosterona es importante para el equilibrio del sodio, el cloruro y el potasio, y también para mantener el volumen corporal total. Para comprender la importancia de la aldosterona, se debe considerar que la hormona controla alrededor del 90% de la función mineralocorticoide de las suprarrenales, no obstante el cortisol también participa en la función mineralocorticoide. Si bien la aldosterona tiene una actividad mineralocorticoide 3.000 veces mayor que el cortisol, la cantidad de cortisol en el suero es casi 2.000 veces la de la aldosterona. Por efecto de la potencia de la aldosterona, resulta crucial que el organismo no tenga un exceso o una insuficiencia de este esteroide potente. Las consecuencias del exceso de aldosterona son la disminución del potasio y la debilidad muscular, en tanto las concentraciones bajas de la hormona inducen elevación del potasio y toxicidad cardíaca.
Glucocorticoides
Las hormonas glucocorticoides, en particular el cortisol, se sintetizan en la zona fascicular y en la zona reticular de la glándula suprarrenal. Las concentraciones en sangre de estas hormonas se encuentran reguladas por mecanismos de retroalimentación negativos del sistema hipotálamo-hipófiso-suprarrenal (HHS; figura 49-10).
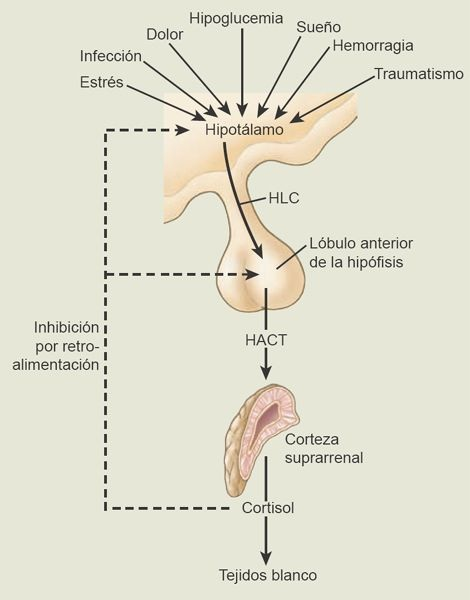
Al igual que otras hormonas hipofisarias reciben control de factores liberadores provenientes del hipotálamo, la hormona liberadora de corticotropina (HLC) es importante para controlar la liberación de HACT. Las concentraciones de cortisol aumentan al tiempo que las de HACT lo hacen y disminuyen cuando la HACT cae. Existe una variación diurna considerable de los niveles de HACT, que alcanzan su máximo temprano por la mañana (entre las 6 a.m. y las 8 a.m.) y declinan al tiempo que el día avanza. Esto parece deberse a una actividad rítmica en el SNC, que genera brotes de secreción de HLC y, a su vez, de secreción de HACT. Este patrón diurno se revierte en personas que trabajan durante la noche y duermen durante el día. El ritmo también puede modificarse con el estrés físico y psicológico, la depresión endógena, la psicosis maniacodepresiva y la hepatopatía u otros padecimientos que afectan el metabolismo del cortisol.
Los glucocorticoides llevan a cabo una función necesaria en respuesta al estrés y resultan esenciales para la sobrevivencia. Cuando se producen como parte de la respuesta al estrés, estas hormonas auxilian en la regulación de las funciones metabólicas del organismo y en el control de la respuesta inflamatoria. Las acciones del cortisol se resumen en la tabla 49-4. Muchas de las acciones anti-inflamatorias que se atribuyen al cortisol se producen con la administración de concentraciones farmacológicas de la hormona.
Defectos metabólicos
El cortisol estimula la síntesis de glucosa en el hígado, promueve la degradación de las proteínas y genera la movilización de los ácidos grasos. Mientras las proteínas corporales se degradan, se movilizan los aminoácidos y transportan hacia el hígado, donde se utilizan para la obtención de glucosa (es decir, gluconeogénesis). La movilización de los ácidos grasos modifica el metabolismo de la célula para dejar de utilizar la glucosa como fuente de energía y comenzar a utilizar los ácidos grasos con este fin. Al tiempo que la producción de glucosa en el hígado aumenta y su consumo periférico cae, se desarrolla una resistencia moderada a la insulina. En las personas con diabetes mellitus y quienes tienden a esta enfermedad, esto tiene el efecto de incrementar la glucemia.
Efectos psicológicos
Las hormonas glucocorticoides parecen estar involucradas en forma directa o indirecta en el comportamiento emocional. En el tejido cerebral se han identificado receptores para estas hormonas, lo que sugiere que desempeñan algún papel en la regulación de la conducta. Se sabe que las personas que reciben tratamiento con hormonas de la corteza suprarrenal muestran conductas variables, que van de poco aberrantes hasta la psicóticas.
Efectos inmunitarios e inflamatorios
El cortisol influye sobre aspectos múltiples de la función inmunitaria y la capacidad de respuesta inflamatoria. Se requieren grandes cantidades de cortisol para obtener una acción antiinflamatoria efectiva. Esto se logra mediante la administración de dosis farmacológicas, más que fisiológicas, de cortisol sintético. El aumento del cortisol bloquea la inflamación en una fase temprana al reducir la permeabilidad capilar y estabilizar las membranas lisosómicas, de tal manera que no se liberan los mediadores inflamatorios. El cortisol suprime la respuesta inmunitaria al limitar la inmunidad humoral y mediada por células. Con esta respuesta inflamatoria menor viene una reducción de la fiebre. Durante la fase de cicatrización, el cortisol suprime la actividad de los fibroblastos y con ello limita la formación de tejido cicatrizal. El cortisol también inhibe la síntesis de prostaglandinas, lo que podría explicar en gran parte de sus acciones antiinflamatorias.
Supresión farmacológica de la función suprarrenal
Un aspecto muy significativo del tratamiento a largo plazo con formulaciones farmacológicas de glucocorticoides es la insuficiencia suprarrenal que se verifica tras suspender la administración de los fármacos. La insuficiencia se debe a la supresión del eje HHS. La supresión crónica induce la atrofia de la glándula suprarrenal, y el retiro abrupto de los medicamentos puede inducir insuficiencia suprarrenal aguda. El período de recuperación para conseguir la normalización de la función suprarrenal puede ser prolongado y requerir hasta 12 meses o más.
Pruebas de función suprarrenal
Es posible recurrir a varias pruebas diagnósticas para evaluar la función de la corteza suprarrenal y del sistema HHS. Las concentraciones sanguíneas de cortisol, aldosterona y HACT pueden medirse mediante técnicas de inmunoensayo. La medición de la excreción de varios productos terminales del metabolismo de las hormonas suprarrenales en una muestra de orina de 24 h aporta información sobre las alteraciones de la biosíntesis de las hormonas de la corteza suprarrenal. El cortisol libre en orina de 24 h, las concentraciones nocturnas séricas o salivales de cortisol (entre las11 p. m. y la medianoche) y la prueba de supresión nocturna con 1 mg de dexametasona son pruebas para detección excelentes en el caso del síndrome de Cushing.
Las pruebas de supresión y estimulación constituyen un medio para valorar la condición del sistema de retroalimentación HHS. Por ejemplo, es posible administrar una dosis de prueba de HACT para valorar la respuesta de la corteza suprarrenal a la estimulación. En forma similar, la administración de dexametasona, un glucocorticoide sintético, constituye un medio para cuantificar la supresión por retroalimentación negativa de la HACT. Los tumores suprarrenales y los tumores en los que se identifica producción ectópica de HACT suelen no responder a la supresión de la HACT con dexametasona. Las pruebas de HLC pueden aplicarse para diagnosticar un tumor hipofisario secretor de HACT (es decir, la enfermedad de Cushing). La estimulación con corticotropina (cosintropina) es la prueba diagnóstica de empleo más frecuente para la valoración de la capacidad de respuesta del eje HHS.
Hiperplasia suprarrenal congénita
La hiperplasia suprarrenal congénita (HSC), o síndrome adrenogenital, describe un trastorno congénito generado por un rasgo autosómico recesivo en que existe una insuficiencia de cualquiera de las enzimas necesarias para la síntesis del cortisol (figura 49-10). Una característica común de todos los tipos de HSC es un defecto de la síntesis de cortisol que tiene como consecuencia el aumento de las concentraciones de HACT y la hiperplasia suprarrenal. El aumento de las concentraciones de HACT estimula en exceso las vías para la síntesis de andrógenos suprarrenales.
Los mineralocorticoides pueden sintetizarse en cantidades excesivas o insuficientes, lo que depende de la insuficiencia enzimática precisa. Se ven afectados neonatos de ambos sexos. En los varones rara vez se establece el diagnóstico al nacer, a menos que presenten aumento del tamaño de los genitales o pérdida de sal, y manifiesten una crisis suprarrenal. En las niñas recién nacidas el incremento de los andrógenos genera un síndrome de virilización, con genitales ambiguos y clitoromegalia, fusión de los labios mayores y presencia de seno urogenital (figura 49-11). En los niños de ambos sexos son normales otras características sexuales secundarias y la fertilidad no se afecta cuando se instituye un tratamiento apropiado.
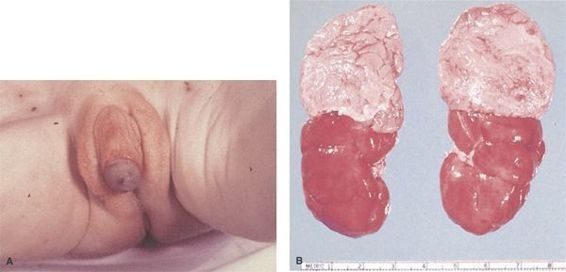
Existe un espectro patológico por insuficiencia de la 21-hidroxilasa, que varía desde la HSC virilizante simple hasta una insuficiencia enzimática completa con pérdida de sal. La HSC virilizante simple compromete la síntesis de colesterol y los productos para síntesis de esteroides se desvían hacia la producción de andrógenos. Las personas con estas insuficiencias suelen sintetizar una cantidad suficiente de aldosterona o intermedios productos intermedios de la aldosterona, que impide la aparición de signos y síntomas de insuficiencia de mineralocorticoides. La variante perdedora de sal se acompaña de una producción insuficiente de aldosterona y sus productos intermedios. Esto da origen a trastornos de líquidos y electrolitos para el quinto día de vida (lo que incluye hiponatremia, vómito, deshidratación y shock). No siempre existe hiperpotasemia, de manera que no debe considerarse un parámetro diagnóstico importante para la detección.
La insuficiencia de 11-β-hidroxilasa es rara y se manifiesta con gravedad diversa. Las personas afectadas tienen una síntesis excesiva de andrógenos y compromiso de la conversión de la 11-desoxicorticoesterona en corticoesterona. La producción excesiva de 11-desoxicorticoesterona, que cuenta con actividad mineralocorticoide, es responsable de la hipertensión que acompaña a esta insuficiencia. El diagnóstico de HSC depende de la evaluación bioquímica precisa de los metabolitos de la vía del cortisol, y de los signos y síntomas clínicos. También resultan invaluables las pruebas genéticas. Sin embargo, la correlación entre el fenotipo y el genotipo no siempre es precisa.
El tratamiento médico de la HSC incluye la restitución con glucocorticoides por vía oral o parenteral. El acetato de fludrocortisona, un mineralocorticoides, también puede administrarse a niños perdedores de sal. Con base en el grado de virilización existe indicación para la cirugía reconstructiva durante los primeros 2 años de vida, con el objetivo de reducir el tamaño del clítoris, separar los labios y permeabilizar la vagina.
Insuficiencia suprarrenocortical
Existen 2 variantes de insuficiencia suprarrenal: la primaria y la secundaria, en la tabla 49-5 se resumen sus características diferenciales. La insuficiencia suprarrenal primaria, o enfermedad de Addison, deriva de la destrucción de la glándula suprarrenal. La insuficiencia suprarrenal secundaria se debe a un trastorno del sistema HHS.
Insuficiencia suprarrenocortical primaria
La enfermedad de Addison hace referencia tan sólo a la insuficiencia suprarrenal primaria en que existe insuficiencia de las hormonas de la corteza suprarrenal y las concentraciones de HACT aumentan por efecto de la falta de inhibición mediante retroalimentación.
Etiología y patogénesis
Esta enfermedad es un trastorno raro, en que se destruyen todas las capas de la corteza suprarrenal. La destrucción autoinmunitaria es la etiología más frecuente de enfermedad de Addison en Estados Unidos. Antes de 1950, la tuberculosis era la causa principal de la enfermedad de Addison en Estados Unidos y Canadá, y aún es una etiología importante en países en que la infección tiene mayor prevalencia. Entre las etiologías infrecuentes se encuentran el carcinoma metastásico, la infección micótica (en particular, histoplasmosis), la infección por citomegalovirus, la enfermedad amiloide y la hemocromatosis. Puede ocurrir hemorragia suprarrenal bilateral en personas que reciben anticoagulantes, durante la cirugía a corazón abierto y durante el parto o con un traumatismo mayor. La insuficiencia suprarrenal puede derivar del sida, en el que la glándula es destruida por distintos fármacos infecciosos oportunistas. Los medicamentos que inhiben la síntesis de los glucocorticoides o inducen su degradación excesiva pueden inducir insuficiencia suprarrenal (ej.ketoconazol).
Manifestaciones clínicas
La corteza suprarrenal tiene una capacidad de reserva grande y las manifestaciones de la insuficiencia no se hacen aparentes sino hasta que cerca del 90% de la glándula se destruye. Estas manifestaciones se relacionan ante todo con la insuficiencia de mineralocorticoides, la insuficiencia de glucocorticoides y la hiperpigmentación que deriva del aumento de las concentraciones de HACT. Si bien la falta de andrógenos suprarrenales (es decir, DHEAS) tiene pocos efectos en el varón debido a que los testículos sintetizan esta hormona, las mujeres muestran disminución del vello axilar y el púbico.
La insuficiencia de mineralocorticoides induce aumento de las pérdidas urinarias de sodio, cloruro y agua, a la par de una disminución de la excreción del potasio (figura 49-12).
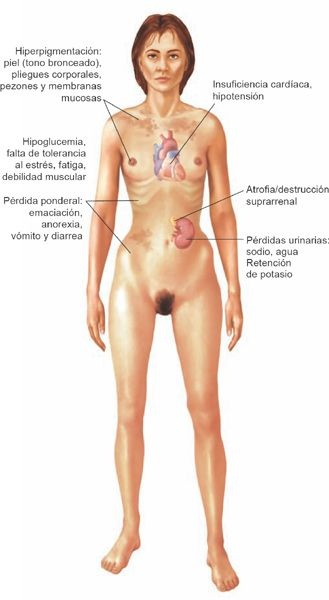
La consecuencia es la hiponatremia, la pérdida del líquido extracelular, la disminución del gasto cardíaco y la hiperpotasemia. Puede existir un deseo anómalo de consumir sal. La hipotensión ortostática es común. La deshidratación, la debilidad y la fatiga son síntomas tempranos frecuentes. Si la pérdida de sodio y agua es extrema, se producen colapso cardiovascular y shock. Por efecto de la carencia de glucocorticoides, la persona con enfermedad de Addison tiene poca tolerancia al estrés. Esta insuficiencia induce hipoglucemia, letargo, debilidad, fiebre y síntomas gastrointestinales, como anorexia, náuseas, vómito y pérdida ponderal.
Se presenta hiperpigmentación por el aumento de las concentraciones de HACT. La piel adquiere un aspecto bronceado tanto en regiones expuestas como no expuestas, y los pliegues y los puntos de presión normales tienden a desarrollar una tonalidad en particular oscura. Las encías y las membranas de la mucosa bucal pueden desarrollar una coloración azulnegruzca. La secuencia de aminoácidos de la HACT es en extremo similar a la de la hormona estimulante de los melanocitos; se presenta hiperpigmentación en más del 90% de los individuos con enfermedad de Addison, y resulta útil para diferenciar las variantes primaria y secundaria de insuficiencia suprarrenal.
Tratamiento
La enfermedad de Addison, al igual que la diabetes mellitus tipo 1, es un trastorno crónico del metabolismo que hace necesario el tratamiento de restitución hormonal de por vida. La regulación cotidiana de la fase crónica de la enfermedad de Addison suele lograrse mediante el tratamiento de restitución oral, con administración de dosis más altas durante los períodos de estrés.
El agente farmacológico que se utiliza debe tener actividad tanto glucocorticoide como mineralocorticoide. Los mineralocorticoides se requieren sólo en la insuficiencia suprarrenal primaria. La hidrocortisona suele ser el fármaco de elección. En los casos leves, puede resultar suficiente la hidrocortisona sola. La fludrocortisona (un mineralocorticoide) se utiliza en individuos que no logran un efecto suficiente de retención de sal con la hidrocortisona. La restitución con DHEAS también puede resultar útil en las mujeres.
Puesto que las personas con el trastorno tienen probabilidad de sufrir episodios de hiponatremia e hipoglucemia, necesitan tener un horario regular para la alimentación y el ejercicio. Las personas con enfermedad de Addison también muestran una capacidad limitada para responder ante las infecciones, los traumatismos y otros factores de estrés. Situaciones de este tipo requieren una atención y un manejo médico inmediato. Debe indicarse a los pacientes con enfermedad de Addison que lleven consigo un brazalete o una placa de alerta médica.
Insuficiencia suprarrenocortical secundaria
La insuficiencia suprarrenal secundaria puede presentarse como consecuencia del hipopituitarismo o por la remoción quirúrgica de la glándula hipófisis. La insuficiencia suprarrenal terciaria deriva de un defecto hipotalámico. Sin embargo, una causa mucho más común que cualquiera de estas patologías es el retiro rápido de los glucocorticoides suministrados con fines terapéuticos para el manejo del asma o alguna exacerbación de la esclerosis múltiple. Estos fármacos suprimen el eje HHS, lo que da origen a una atrofia de la corteza suprarrenal y a la pérdida de la síntesis de cortisol.
Esta supresión persiste durante un período prolongado tras la suspensión del tratamiento y puede resultar crítica durante los períodos de estrés o cuando se practica una cirugía.
Crisis suprarrenal aguda
La crisis suprarrenal aguda es una situación que pone en riesgo la vida. Si la enfermedad de Addison es el problema subyacente, la exposición incluso a enfermedades o tensiones menores puede precipitar náuseas, vómito, debilidad muscular, hipotensión, deshidratación y colapso vascular. El desarrollo de una crisis suprarrenal puede ser súbito o instaurarse a lo largo de varios días. Los síntomas pueden ocurrir de manera súbita en los niños con variantes de pérdida de sal de HSC. La hemorragia suprarrenal bilateral masiva induce una variante fulminante aguda de insuficiencia suprarrenal. La hemorragia puede derivar de una septicemia meningocócica, un traumatismo suprarrenal, el tratamiento con anticoagulantes, la trombosis de la vena suprarrenal o las metástasis a glándulas suprarrenales.
La insuficiencia suprarrenal se maneja con tratamiento de restitución hormonal, que incluye una combinación de glucocorticoides y mineralocorticoides. En la insuficiencia suprarrenal aguda deben seguirse de las 5 «S» para el manejo:
- sal para restitución,
- suplementación de azúcar (dextrosa),
- suplementación de esteroides,
- soporte del desempeño fisiológico, y
- sondeo para identificación y tratamiento de la causa subyacente (ej. infección).
El volumen de líquido extracelular debe restituirse, con varios litros de solución salina al 0,9% y dextrosa al 5%. La restitución con glucocorticoides se logra por medio de la administración intravenosa ya sea de dexametasona o hidrocortisona.
La dexametasona se prefiere en el período agudo por 2 razones: tiene acción prolongada (12 h a 24 h) y no interfiere con la medición de los esteroides séricos o urinarios durante las pruebas de estimulación subsecuentes con corticotropina (HACT) si se necesita establecer el diagnóstico.
En forma posterior se administra hidrocortisona, ya sea por vía intravenosa o intramuscular, a intervalos de 6 h, y luego se reduce la dosis en el transcurso de uno a 3 días hasta los niveles de mantenimiento. El tratamiento de restitución con hidrocortisona por vía oral puede reiniciarse una vez que se suspende la infusión de solución salina y la persona es capaz de consumir alimentos y líquidos por vía oral. No se requiere tratamiento con mineralocorticoides cuando se administran dosis altas de hidrocortisona, pero al tiempo que la dosis se reduce suele ser necesario agregar fludrocortisona. El tratamiento de restitución con glucocorticoides y mineralocorticoides se vigila a partir de la medición de la frecuencia cardíaca y la presión arterial, la vigilancia de los valores de los electrolitos séricos, y el ajuste de la actividad de la renina plasmática hasta alcanzar un valor alto en el intervalo normal.
Exceso de hormonas glucocorticoides (síndrome de Cushing)
El término síndrome de Cushing se refiere a las manifestaciones del hipercortisolismo de cualquier causa. Tres variantes relevantes del síndrome de Cushing derivan de una síntesis excesiva de glucocorticoides en el organismo. Una es la variante hipofisaria, que se debe a la producción excesiva de HACT en un tumor hipofisario. Esta variante de enfermedad fue la descrita en su origen por Cushing. Por ende, se denomina enfermedad de Cushing. La segunda variante es la suprarrenal, que deriva de un tumor suprarrenal benigno o maligno. La tercera variante es el síndrome de Cushing ectópico, que depende de un tumor no hipofisario secretor de HACT. Ciertos tumores malignos que se desarrollan fuera de la hipófisis, como el carcinoma de células pequeñas del pulmón, pueden secretar HACT o, rara vez, HLC, e inducir síndrome de Cushing. El síndrome de Cushing también puede producirse por el tratamiento a largo plazo con formulaciones farmacológicas potentes de glucocorticoides; esta variante se denomina síndrome de Cushing iatrógeno.
Manifestaciones clínicas
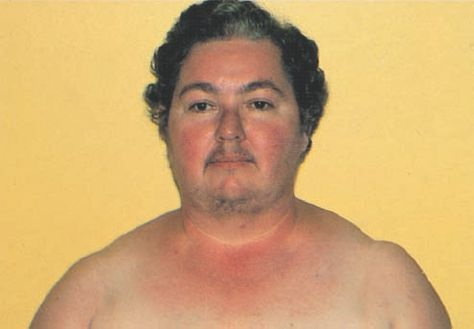
Las manifestaciones principales del síndrome de Cushing representan una exageración de las acciones del cortisol (tabla 49-4). Las anomalías del metabolismo de los lípidos induce un depósito peculiar de las grasas, que se caracteriza por generar protrusión abdominal, acumulación de cojinetes adiposos en la región alta de la espalda o «giba de búfalo», y «cara de luna llena», en que la cara adquiere un aspecto redondo y pletórico (figuras 49-13 y 49-14). Existe debilidad muscular y las extremidades son delgadas por efecto de la degradación de las proteínas y el desgaste muscular.
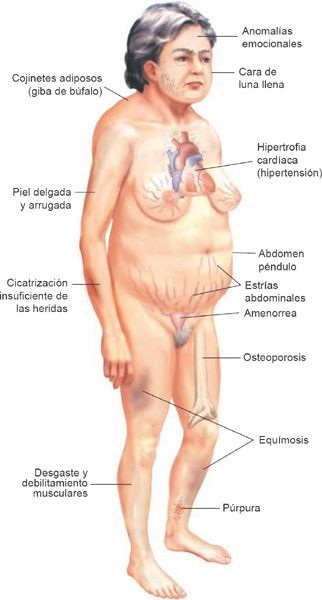
En los casos avanzados, la piel que cubre los antebrazos y las piernas se adelgaza, y adquiere un aspecto apergaminado. Estrías purpúricas, o cicatrices por distensión, que derivan del estiramiento de la piel y los tejidos subcutáneos con debilitamiento catabólico, se distribuyen sobre las mamas, la región proximal de los muslos y el abdomen. Puede desarrollarse osteoporosis como consecuencia de la destrucción de las proteínas del hueso y las alteraciones del metabolismo del calcio, lo que genera dorsalgia, fracturas vertebrales por compresión y fracturas costales. Al tiempo que el calcio se moviliza a partir del hueso pueden desarrollarse cálculos renales.
Los glucocorticoides poseen propiedades mineralocorticoides. Esto genera hipopotasemia, como consecuencia de una excreción excesiva de potasio, e hipertensión, que deriva de la retención del sodio. Las respuestas inflamatoria e inmunitaria se encuentran inhibidas, lo que da origen a una mayor susceptibilidad a la infección. El cortisol intensifica la secreción de ácido gástrico, lo que puede provocar ulceración gástrica y hemorragia. Un incremento concurrente de las concentraciones de andrógenos induce hirsutismo, acné leve e irregularidad menstrual en la mujer. Las concentraciones excesivas de glucocorticoides pueden dar origen a una labilidad emocional extrema, que va desde la euforia leve y la ausencia de fatiga normal hasta una conducta psicótica franca.
Diagnóstico y tratamiento
El diagnóstico del síndrome de Cushing depende del hallazgo de hipersecreción de cortisol. La determinación de la excreción de cortisol en orina de 24 h constituye un índice confiable y práctico de la secreción de cortisol. Una de las características prominentes del síndrome de Cushing es la pérdida del patrón diurno de secreción del cortisol. La prueba de supresión nocturna con 1 mg de dexametasona también se utiliza como instrumento para detección en el síndrome de Cushing.
Otra prueba es la medición de las concentraciones plasmáticas de HACT. Las concentraciones de HACT deben ser normales o altas en el síndrome de Cushing dependiente de HACT (enfermedad de Cushing y secreción ectópica de HACT), y bajas en el síndrome de Cushing independiente de HACT (tumores suprarrenales). Se realizan distintas pruebas de supresión o estimulación del sistema HHS para Definir con más detalle la causa. Los estudios de IRM o TC aportan un medio para localizar los tumores suprarrenales o hipofisarios. Si no recibe tratamiento, el síndrome de Cushing induce morbilidad grave e incluso la muerte.
La elección de tratamiento quirúrgico, radioterapia o manejo farmacológico se determina en gran medida a partir de la causa del hipercortisolismo. La meta del tratamiento del síndrome de Cushing es eliminar o corregir la fuente del hipercortisolismo sin generar daño permanente a la hipófisis o las suprarrenales. La extirpación transesfenoidal de un adenoma hipofisario o una hemihipofisectomía constituyen el método preferido para el tratamiento de la enfermedad de Cushing. Esto permite retirar tan sólo el tumor y no toda la glándula hipófisis. Tras una extirpación exitosa, la persona debe recibirtratamiento de restitución con cortisol durante 6 a 12 meses o hasta que se recupere la función suprarrenal. Las personas también pueden recibir radioterapia hipofisaria, pero los efectos completos del tratamiento pudieran no alcanzarse durante 3 a 12 meses. Puede recurrirse a la suprarrenalectomía unilateral o bilateral cuando existe un adenoma suprarrenal. De ser posible, también se extirpan los tumores con producción ectópica de HACT. Los fármacos farmacológicos que bloquean la síntesis de esteroides (ej. mitotano, ketoconazol y metirapona) pueden utilizarse para manejar a las personas con tumores con producción ectópica o carcinoma suprarrenales irresecables. Muchos de estos individuos también requieren profilaxis contra la neumonía por Pneumocystis jiroveci (antes Pneumocystis carinii) por efecto de la inmunosupresión profunda que inducen las concentraciones excesivas de glucocorticoides.
Masa suprarrenal de identificación incidental
El «incidentaloma» es una masa que se identifica de manera inesperada en una glándula suprarrenal cuando se realiza un procedimiento de imagenología (por otras causas), la mayoría de las veces una TC (pero también una IRM o una ecografía). Los incidentalomas también pueden identificarse en otros órganos (ej. hipófisis, tiroides). Los 2 puntos más importantes por Definir en relación con estos tumores son si se trata de lesiones malignas y si tienen actividad hormonal.
El carcinoma suprarrenal primario es bastante raro, pero otros cánceres, en particular los de pulmón, generan metástasis con frecuencia hacia la glándula suprarrenal (otros cánceres que lo hacen son los de mama, estómago, páncreas, colon, riñón, los melanomas y los linfomas). El tamaño y las características de imagen de la masa pudieran ayudar a determinar si el tumor es benigno o maligno.
Un procedimiento para detección apropiado para excluir una lesión con actividad hormonal incluye la realización de pruebas para descartar feocromocitoma, síndrome de Cushing y síndrome de Conn (exceso de mineralocorticoides).