02. Hipófisis y trastornos del crecimiento
La glándula hipófisis, o pituitaria, tiene el tamaño de un chícharo y se ubica en la base del cerebro, donde se aloja en la depresión con forma de silla de montar en el hueso esfenoides que se denomina silla turca. Un tallo corto con forma de embudo, el infundíbulo, conecta la glándula hipófisis con el hipotálamo. La glándula hipófisis cuenta con 2 componentes: un lóbulo posterior (neurohipófisis), o componente neural, y un lóbulo anterior (adenohipófisis), o componente glandular.
El lóbulo anterior de la glándula hipófisis sintetiza HACT, hormona estimulante de la tiroides (HET), hormona del crecimiento (HC), hormonas gonadotrópicas (hormona estimulante del folículo [HEF] y hormona luteinizante [HL]) y prolactina. Cuatro de ellas, HACT, HET, HL y HEF, controlan la secreción de hormonas a partir de otras glándulas endocrinas:
- La HACT controla la liberación de cortisol a partir de la glándula suprarrenal.
- La HET controla la secreción de hormonas tiroideas a partir de la glándula tiroides.
- La HL regula las hormonas sexuales.
- La HEF regula la fertilidad.
Valoración de la función hipotálamo-hipofisaria
La valoración de la función hipotálamo-hipófisis ha sido posible gracias al desarrollo de muchas técnicas nuevas de imagenología y radioinmunoensayo. La valoración de la condición basal de las hormonas de las células blanco hipotálamo-hipofisarias implica la medición de las sustancias siguientes (de manera idónea, los especímenes de laboratorio deben obtenerse antes de las 8 a. m.):
- Cortisol sérico.
- Prolactina sérica.
- Tiroxina y HET séricas.
- Testosterona sérica (varón) o estrógenos séricos (mujeres) y HL/HEF séricas.
- HC/factor de crecimiento similar a la insulina tipo 1 (FCI-1) séricas.
- Osmolalidad plasmática y osmolalidad urinaria.
Los estudios de imagenología (ej. imágenes de resonancia magnética nuclear [IRM] del hipotálamo y la hipófisis) también deben realizarse según se requiera. Cuando se precisa información adicional relativa a la función hipofisaria, se solicitan pruebas combinadas de función del hipotálamo-hipófisis. Estas pruebas consisten ante todo de pruebas de estimulación hormonales (ej. prueba rápida de estimulación con HACT) o pruebas de supresión (ej. prueba de supresión de HC).
Suele ser importante analizar la función hipofisaria, en particular si se descubren adenomas hipofisarios y se considera recurrir a la cirugía o la radioterapia. Las técnicas diagnósticas incluyen tanto pruebas estáticas como dinámicas, así como una valoración radiológica según se requiera.Cualesquiera de los sistemas mencionados antes pueden verse afectados, ya sea por la insuficiencia o el exceso de las hormonas que secretan con normalidad.
Tumores hipofisarios
Los tumores hipofisarios pueden dividirse en primarios y secundarios (es decir, lesiones metastásicas). Los tumores de la hipófisis pueden clasificarse además como tumores funcionales que secretan hormonas hipofisarias y tumores no funcionales que no secretan hormonas. Pueden variar en tamaño desde lesiones pequeñas que no aumentan el tamaño de la glándula (microadenomas, <10 mm) hasta tumores grandes y expansivos (macroadenomas, >10 mm) que erosionan la silla turca y ejercen presión sobre las estructuras craneales circundantes. Los tumores pequeños no funcionales se identifican hasta en el 20% de las autopsias de adultos. Los adenomas benignos constituyen la mayor parte de los tumores funcionales del lóbulo anterior de la hipófisis. Los carcinomas hipofisarios son menos comunes. Los adenomas funcionales puede subdividirse de acuerdo con el tipo celular y el tipo de hormona secretada (tabla 49-1).
Hipopituitarismo
El hipopituitarismo, que se caracteriza por una disminución de la secreción de hormonas hipofisarias, es una condición que afecta a muchos otros sistemas endocrinos. Su etiología puede ser congénita o puede derivar de distintas anomalías adquiridas (recuadro 49-1). El hipopituitarismo se relaciona con un aumento de la morbilidad y la mortalidad.
Recuadro 49-1. Causas de hipopituitarismo:
- Tumores y lesiones con efecto de masa: adenomas hipofisarios, quistes, cáncer metastásico y otras lesiones.
- Cirugía o radiación hipofisaria.
- Lesiones infiltrativas e infecciones: hemocromatosis, hipofisitis linfocítica.
- Infarto hipofisario: infarto de la glándula hipófisis tras una pérdida hemática sustancial durante el parto (síndrome de Sheehan).
- Apoplejía hipofisaria: hemorragia súbita en la glándula hipófisis.
- Trastornos genéticos: defectos congénitos infrecuentes que afectan a una o más hormonas hipofisarias.
- Síndrome de silla turca vacía: aumento del volumen de las silla turca, que no se encuentra ocupada del todo por tejido hipofisario.
- Trastornos hipotalámicos: tumores y lesiones con efecto de masa (ej. craneofaringiomas y neoplasias metastásicas), radiación hipotalámica, lesiones infiltrativas (ej. sarcoidosis), traumatismo, infecciones.
De manera característica, del 70% al 90% del lóbulo anterior de la hipófisis debe destruirse antes de que exista evidencia clínica de hipopituitarismo. Las manifestaciones clínicas del hipopituitarismo suelen desarrollarse de manera gradual, pero puede revelarse como una condición aguda y que pone en riesgo la vida. Por lo general, las personas refieren un malestar crónico, con debilidad, fatiga, pérdida del apetito, disfunción sexual e intolerancia al frío. Sin embargo, la insuficiencia de HACT (insuficiencia suprarrenal secundaria) es la insuficiencia endocrina más grave, que da origen a debilidad, náuseas, anorexia, fiebre e hipotensión postural.
La pérdida de las hormonas del lóbulo anterior de la hipófisis tiende a seguir una secuencia típica, en particular con pérdida progresiva de la reserva hipofisaria secundaria a tumores o radioterapia hipofisaria previa. La secuencia de pérdida de hormonas hipofisarias puede recordarse mediante la mnemotecnia «Go Look For The Adenoma» (busque el adenoma):
- HC: de manera característica, la secreción de HC es la primera que se pierde (la G corresponde a grouth hormone).
- HL: induce insuficiencia de hormonas sexuales.
- HEF: induce infertilidad.
- HET: conduce al hipotiroidismo secundario.
- HACT: por lo general, es la última en desarrollar insuficiencia y generar la insuficiencia suprarrenal secundaria.
El tratamiento del hipopituitarismo incluye el manejo de cualquier causa subyacente identificada. Las insuficiencias hormonales deben controlarse según, se requiera, tomando como base las concentraciones iniciales de hormonas y en realización de pruebas hipofisarias más sofisticadas cuando resulte apropiado. La restitución de cortisol se inicia una vez que existe insuficiencia de HACT; la restitución tiroidea cuando se detecta insuficiencia de HET; la restitución de hormonas sexuales cuando existe insuficiencia de HL y HEF. La restitución de HC se encuentra indicada cuando existe insuficiencia en pacientes pediátricos, y se utiliza cada vez más para el tratamiento de la insuficiencia de la hormona en los adultos.
Crecimiento y trastornos de la hormona del crecimiento
Varias hormonas resultan esenciales para el crecimiento corporal normal y su maduración, lo que incluye a HC, insulina, hormonas tiroideas y andrógenos. Además de sus acciones sobre el metabolismo de carbohidratos y grasas, la insulina desempeña un papel esencial sobre los procesos de crecimiento. Los niños con diabetes, en especial los que presentan dificultad para equilibrar su glucemia, con frecuencia no pueden crecer con normalidad incluso si sus concentraciones de HC son normales. Cuando las concentraciones de las hormonas tiroideas son menores que las normales, el crecimiento óseo y el cierre epifisario se retrasan. Los andrógenos, como la testosterona y la dihidrotestosterona, ejercen efectos anabólicos de crecimiento por medio de sus acciones sobre la síntesis de proteínas. Los glucocorticoides en concentraciones excesivas inhiben el crecimiento, en apariencia debido a su efecto antagónico sobre la secreción de la HC.
Hormona del crecimiento
La hormona del crecimiento (HC), también denominada somatotropina, es una hormona polipeptídica con 191 aminoácidos que se sintetiza y secreta en células especiales en el lóbulo anterior de la hipófisis, que se denominan somatotropos. Han sido una creencia común que la HC se sintetizaba ante todo durante los períodos de crecimiento. Sin embargo, se ha probado que esto es incorrecto, puesto que la tasa de síntesis de HC en adultos es casi tan alta como en los niños. La HC es necesaria para el crecimiento y contribuye a la regulación de las funciones metabólicas (figura 49-1).
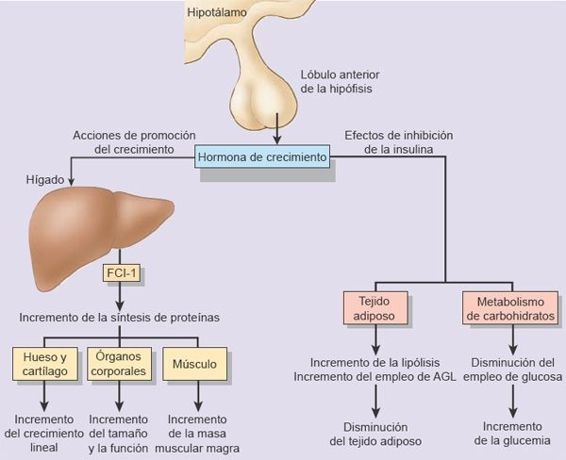
La HC estimula todos los aspectos del crecimiento del cartílago. Uno de los efectos más evidentes de la HC ocurre sobre el crecimiento lineal del hueso, que deriva de su acción sobre las placas de crecimiento epifisarias en los huesos largos. El grosor del hueso se incrementa por el aumento del crecimiento perióstico. Los órganos viscerales y endocrinos, el músculo esquelético y el cardíaco, la piel y tejido conectivo muestran todos un mayor crecimiento en respuesta a la HC.
Además, la HC promueve la velocidad de síntesis de proteínas en el organismo. Favorece la movilización de los ácidos grasos e incrementa su utilización para la obtención de combustible y mantiene o incrementa las concentraciones de glucosa en la sangre al disminuir su consumo como combustible. La HC tiene un efecto inicial de aumento de las concentraciones de insulina. Sin embargo, el efecto predominante del exceso persistente de la HC es el incremento las concentraciones de glucosa a pesar de un aumento de la insulina. Esto se debe a que la HC induce resistencia a la insulina en los tejidos periféricos, lo que inhibe la captación de glucosa en los tejidos muscular y adiposo.
Muchos efectos de la HC dependen de los factores de crecimiento similares a la insulina (FCI), también denominados somatomedinas, que se sintetizan de manera principal en el hígado. La HC no puede generar en forma directa crecimiento óseo. En vez de esto, actúa por vía indirecta al inducir al hígado a producir FCI. Estos péptidos FCI actúan sobre el cartílago y el hueso para promover su crecimiento. Se han identificado por lo menos 4 FCI. De estos, el FCI-1 (somatomedina C) parece ser el más importante desde la perspectiva del crecimiento. También es el FCI que se mide con más frecuencia en las pruebas de laboratorio. Los FCI se han secuenciado y tienen estructuras similares a las de la proinsulina. Esto explica la actividad similar a la insulina de los FCI y la acción débil de esa hormona sobre el crecimiento. Las concentraciones de FCI son en sí mismas influidas por una familia de 6 factores de unión denominados proteínas de unión a FCI (PUFCI).
La HC se transporta libre en el plasma y tiene una vida media aproximada de 20 min a 50 min.
Dos hormonas hipotalámicas regulan la secreción de HC:
- Hormona liberadora de la HC (HLHC), que incrementa la liberación de la hormona.
- Somatostatina, que inhibe la liberación de HC.
Una tercera hormona, la recién identificada grelina, también tiene un papel importante, que consiste en incrementar el apetito al estimular al hipotálamo y también en aumentar la acumulación de lípidos en el tejido adiposo visceral en el abdomen.
Factores neurales, metabólicos y hormonales regulan en forma estrecha estos factores de influencia hipotalámicos (es decir, HLHC y somatostatina). Por ejemplo, un estudio conducido en personas obesas encontró concentraciones bajas de grelina, debido a que se absorbía en el tejido adiposo del abdomen y no era cuantificable en el suero. Por ende, se observó una mayor morbilidad y mortalidad en estas personas obesas. Las 2 afecciones comórbidas más frecuentes en las personas obesas fueron hipertensión y diabetes mellitus tipo 2.
La secreción de HC fluctúa en un período de 24 h, para alcanzar concentraciones máximas entre 1 h y 4 h después del inicio del sueño (es decir, durante las fases 3 y 4 del sueño). A la secreción de HC la estimulan la hipoglucemia, el ayuno, la inanición, las concentraciones elevadas de aminoácidos en la sangre (en particular, arginina) y condiciones de estrés como traumatismos, excitación, tensión emocional y ejercicio intenso. La HC se inhibe ante el incremento de las concentraciones de glucosa, la liberación de ácidos grasos libres, el cortisol y la obesidad.
Niños de peso bajo
El peso bajo es una condición en la que la talla que se alcanza se encuentra por debajo del tercer percentil, o el crecimiento lineal es inferior al normal para la edad y el sexo. El peso bajo, o retraso del crecimiento, tiene distintas causas, como anomalías cromosómicas como el síndrome de Turner, la insuficiencia de HC, el hipotiroidismo y el panhipopituitarismo (es decir, insuficiencia de todas las hormonas hipofisarias). Otras condiciones que se sabe inducen peso bajo incluyen la desnutrición proteicocalórica, trastornos crónicos como la nefropatía crónica y la diabetes mellitus mal controlada, los síndromes de malabsorción, y ciertos tratamientos como la administración excesiva de glucocorticoides. Los trastornos emocionales también pueden inducir al desarrollo de trastornos endocrinos funcionales y generar enanismo psicosocial. Las causas de peso bajo se resumen en el recuadro 49-2.
Recuadro 49-2. Causas de peso bajo:
- Variantes normales
- Peso bajo genético o «familiar».
- Peso bajo constitucional.
- Peso bajo al nacer (ej. retraso del crecimiento intrauterino)
- Trastornos endocrinos
- Insuficiencia de hormona del crecimiento (HC).
- Insuficiencia primaria de HC.
- Insuficiencia idiopática de HC.
- Agenesia hipofisaria.
- Insuficiencia secundaria de HC (panhipopituitarismo).
- Producción de HC sin actividad biológica.
- Respuesta deficiente de FCI-1 a concentraciones normales o altas de HC (enanismo tipo Laron).
- Hipotiroidismo.
- Diabetes mellitus mal controlada.
- Exceso de glucocorticoides.
- Endógeno (síndrome de Cushing).
- Exógeno (tratamiento con fármacos glucocorticoides).
- Metabolismo mineral anómalo (ej. seudohipoparatiroidismo).
- Enfermedad crónica y desnutrición
- Enfermedad orgánica o sistémica crónica (ej. asma, en particular cuando se recibe tratamiento con glucocorticoides; cardiopatía o nefropatía).
- Deprivación nutricional.
- Síndrome de malabsorción (ej. esprúe celíaco).
- Trastornos endocrinos funcionales (enanismo psicosocial)
- Trastornos cromosómicos (ej. síndrome de Turner)
- Anomalías esqueléticas (ej. acondroplasia)
La medición precisa de la talla es una parte importante de la exploración física en los niños. Resulta esencial la integración de los antecedentes de desarrollo y el registro en tablas de crecimiento. También son necesarias las curvas de crecimiento y los estudios de velocidad de crecimiento. El diagnóstico de peso bajo no se establece con una sola medición, si no se basa en mediciones secuenciales de la talla, en la velocidad del crecimiento y la talla de los progenitores.
Los procedimientos para el diagnóstico de peso bajo incluyen pruebas para excluir etiologías distintas a las endocrinas. Si la causa es hormonal, se inician procedimientos de análisis hormonales extensos. Por lo general, se determinan los niveles de HC e FCI-1. Pueden realizarse pruebas utilizando insulina (para inducir hipoglucemia), HLHC, levodopa y arginina, todas las cuales estimulan la secreción de HC, de tal manera que sea posible evaluar su reserva. Debido a que la administración de fármacos farmacológicos puede inducir resultados negativos falsos, suelen realizarse 2 pruebas o más para asegurar la precisión. Si se obtiene una elevación rápida de la HC, se considera que el niño es normal. También pueden realizarse pruebas fisiológicas de la reserva de HC (ej. respuesta de la HC al ejercicio). Las concentraciones de FCI-1 suelen ser un reflejo de las de HC, y pueden utilizarse como indicadores de insuficiencia de HC. Las radiografías simples se utilizan para valorar la edad ósea, que la mayoría de las veces muestra retraso. Se recomienda la IRM del área del hipotálamo y la hipófisis en caso de sospecha clínica de lesión.
Una vez que se determina la causa del peso bajo, puede iniciarse el tratamiento. El crecimiento de recuperación es un concepto que se utiliza para describir una velocidad de crecimiento elevada anómala que se presenta al tiempo que el niño se aproxima a la talla normal para la edad. Ocurre tras el inicio del tratamiento para la insuficiencia de HC y el hipotiroidismo, así como tras la corrección de las enfermedades crónicas.
Peso bajo genético y constitucional
Dos variantes de peso bajo, el peso bajo genético y el peso bajo constitucional, no son condiciones patológicas sino variaciones de las directrices poblacionales.
Los niños con peso bajo genético tienden a mostrar una proporción corporal apropiada y tener una talla cercana al promedio de la talla de los progenitores. Entre los niños normales el 95% tiene una talla con una variación de 8 cm (es decir, ±2 desviaciones estándares) del promedio de la talla de los progenitores.
Peso bajo constitucional es un concepto que se aplica para describir a los niños (en particular varones) con peso bajo moderado, constitución delgada y maduración esquelética y sexual tardía, con ausencia de otras causas que expliquen el crecimiento menor.
Enanismo psicosocial
El enanismo psicosocial implica un hipopituitarismo funcional, y se identifica en algunos niños con deprivación emocional. Estos niños suelen presentarse con crecimiento deficiente, abdomen prominente y hábitos deficientes de consumo de alimentos y bebidas. De manera característica, existe el antecedente de alteraciones de las relaciones familiares, en las que el niño ha sufrido abandono o medidas disciplinarias severas. Con frecuencia, el abandono se limita a uno de los niños de la familia. La función de la HC suele recuperar la normalidad una vez que el niño se retira del ambiente limitante. El pronóstico depende del mejoramiento de la conducta y delcrecimiento de recuperación.
Insuficiencia de hormona del crecimiento en niños
Existen distintas variantes de insuficiencia de HC que se presentan durante la niñez. Los niños con insuficiencia idiopática de HC carecen de HLHC hipotalámica, pero sus somatotropos son adecuados, en tanto los niños con tumores hipofisarios o agenesia hipofisaria carecen de somatotropos. El término panhipopituitarismo hace referencia a una condición que induce una insuficiencia de todas las hormonas del lóbulo anterior de la hipófisis. En una condición infrecuente denominada enanismo tipo Laron, las concentraciones de HC son normales o muestran elevación, pero existe un defecto hereditario de la síntesis de FCI que puede manejarse de manera directa mediante restitución con FCI-1.
La insuficiencia congénita de la HC se relaciona con una disminución de la talla al nacer, seguida por una reducción de la velocidad del crecimiento que puede identificarse con la medición cuidadosa durante el primer año de vida, que se hace evidente entre los 1 y 2 años de edad. Las personas con insuficiencia clásica de HC presentan inteligencia normal, peso bajo, obesidad con características faciales inmaduras y cierto retraso de la maduración esquelética. La pubertad con frecuencia se retrasa y los varones con el trastorno presentan microfalo (pene pequeño y anómalo), en particular si la condición se acompaña de insuficiencia de hormona liberadora de gonadotropinas (HLGn). En el neonato, la insuficiencia de HC puede inducir hipoglucemia y convulsiones. Si también existe insuficiencia de HACT, la hipoglucemia con frecuencia es más intensa. La insuficiencia adquirida de HC se desarrolla en una fase posterior de la niñez. Puede derivar de un tumor hipotalámico-hipofisario, en particular si se acompaña de otras insuficiencias de hormonas hipofisarias.
Cuando el peso bajo se debe a una insuficiencia de HC, el tratamiento de elección es el de restitución de la hormona. La HC es específica para la especie, por lo que en el humano sólo resulta efectiva la HC humana. La HC se obtenía antes de hipófisis de cadáveres humanos, pero en la actualidad se produce mediante tecnología de ADN recombinante y se encuentra disponible en cantidad suficiente. La HC se administra a diario mediante inyección subcutánea durante el período de crecimiento activo y puede continuarse su administración hasta la edad adulta.
Los niños con peso bajo por síndrome de Turner e insuficiencia renal crónica también reciben tratamiento con HC. El tratamieto con HC puede valorarse en niños con peso bajo pero sin insuficiencia de HC. La evidencia sugiere que el tratamiento a corto plazo con HC incrementa la velocidad de crecimiento en estos niños. Si bien el efecto de la HC sobre la talla a final no es intenso, puede traer consigo un mejoramiento del bienestar psicológico. Existen inquietudes en torno al consumo inapropiado del medicamento para inducir un crecimiento adicional en niños con función normal de la HC que tienen una talla casi normal. Las directrices para el consumo de la hormona aún deben definirse.
Insuficiencia de hormona del crecimiento en adultos
Existen 2 categorías de insuficiencia de HC en adultos:
- insuficiencia de HC existente durante la niñez.
- insuficiencia de HC desarrollada durante la edad adulta, de manera principal como consecuencia de un hipopituitarismo derivado de un tumor hipofisario o su tratamiento.
Las concentraciones de la HC también pueden declinar con el envejecimiento y ha existido interés en los efectos de las concentraciones decrecientes de la HC en los adultos mayores (que se describencomo somatopausia). La restitución de la HC tiene importancia evidente en el niño en crecimiento. Sin embargo, se está valorando su papel en los adultos (en especial para la somatopausia). Otras de las diferencias entre la insuficiencia de HC durante la niñez y de inicio de la adultez se describen en la tabla 49-2.
La evidencia muestra que la mortalidad cardiovascular aumenta en los adultos con insuficiencia de HC. Se refiere una prevalencia mayor de placas ateroescleróticas y disfunción endotelial tanto en la insuficiencia de HC de la niñez y del adulto. El síndrome por insuficiencia de HC se asocia a una serie de factores de riesgo cardiovascular, que incluyen distribución central del tejido adiposo (que se relaciona con aumento de la grasa visceral), resistencia la insulina y dislipidemia. Estas características también se relacionan con el síndrome metabólico. Además de los factores conocidos como tradicionales para el riesgo cardiovascular, tienden a existir algunos factores no tradicionales de riesgo cardiovascular (ej. elevación de la proteína C reactiva de sensibilidad alta -PCRsa-, que es un marcador de la vía inflamatoria). El tratamiento con HC pueden limitar el impacto potencial de muchos de estos factores de riesgo.
El diagnóstico de la insuficiencia de HC en el adulto se establece al identificar respuestas subnormales de la HC sérica ante los estímulos inductores. Una concentración baja de FCI-1 en presencia de algún trastorno hipofisario conocido también es indicadora de insuficiencia de HC. La medición de las concentraciones basales de HC no permite distinguir de manera confiable entre la secreción de HC normal y subnormal en los adultos. La hipoglucemia inducida con insulina constituye el estándar de oro entre las pruebas para la valoración de la reserva de HC. La prueba de arginina con HLHC quizá sea el estudio que le sigue en utilidad. Otras pruebas de estimulación implican el empleo de arginina, levodopa, clonidina (un agonista α-adrenérgico), glucagón o HLHC sola.
El tratamiento para restitución de HC puede conducir a un incremento de la masa corporal magra y a una disminución de la masa adiposa, al incremento de la densidad mineral ósea, al aumento de la tasa de filtración glomerular, a la disminución de las concentraciones lipídicas, al incremento de la capacidad para el ejercicio y a un mejoramiento de la sensación de bienestar en los adultos con insuficiencia de HC. Los efectos colaterales más frecuentes del tratamiento con HC en los adultos con hipopituitarismo son el edema periférico, las artralgias y las mialgias, el síndrome del túnel del carpo, las parestesias y la disminución de la tolerancia a la glucosa. Los efectos colaterales parecen ser más frecuentes en individuos mayores, más pesados y con tratamiento excesivo, según se juzga a partir de una concentración elevada de FCI-1 en el suero durante el tratamiento.
Talla alta en niños
Justo como existe niños con peso bajo para la edad y el sexo, también existen niños que son altos para su talla y sexo. La talla superior al 95° percentil se considera talla alta. Las variantes normales de la talla alta incluyen la talla alta genética y la talla alta constitucional. Los niños con padres en extremo altos tienden a ser más altos que los niños con padres más pequeños. El concepto de talla alta constitucional se utiliza para describir a un niño que es más alto que sus compañeros y crece a una velocidad que se encuentra dentro del intervalo normal para la edad ósea. Otras causas de talla alta son los trastornos genéticos o cromosómicos, como el síndrome de Marfan o el síndrome xyy.
Los niños excepcionalmente altos (es decir, con talla alta genética y talla alta constitucional) pueden recibir tratamiento con hormonas sexuales—estrógenos en niñas y testosterona en varones— para inducir un cierre epifisario temprano. Este tipo de tratamiento sólo se realiza después de tomar en consideración todos los riesgos implicados. Para que sea efectivo, este tratamiento deben instituirse entre 3 y 4 años antes de la fusión epifisaria esperada.
Exceso de hormona del crecimiento en niños
El exceso de HC que se presenta antes de la pubertad y la fusión de las epífisis de los huesos largos da origen al gigantismo. La secreción excesiva de HC a partir de los adenomas somatotropos induce gigantismo en el niño prepúber. Ocurre cuando las epífisis no se han fusionado y los niveles altos de FCI-1 estimulan el crecimiento esquelético excesivo. Por fortuna, la condición es rara como consecuencia del reconocimiento y tratamiento tempranos del adenoma.
Exceso de hormona del crecimiento en adultos
Cuando se presenta un exceso de HC en la edad adulta o una vez que las epífisis de los huesos largos se cierran, la enfermedad se denomina acromegalia. La incidencia anual de la acromegalia es de 3 a 4 casos por cada millón de personas, con una edad promedio de 40 a 45 años en el momento del diagnóstico.
Etiología y patogénesis
La acromegalia deriva de niveles excesivos de HC, que estimula la secreción hepática de FCI-1, mismo que determina la mayor parte de las manifestaciones clínicas de la acromegalia. La etiología más frecuente (95%) de la acromegalia es el adenoma somatotropo.
Alrededor del 75% de las personas con acromegalia cuentan con un macroadenomas somatotropos en el momento del diagnóstico y casi todo el resto presenta microadenomas. Las otras causas de la acromegalia (<5%) son la secreción excesiva de HLHC a partir de tumores hipotalámicos, la secreción ectópica de HLHC a partir de tumores no endocrinos como los carcinoides o los cáncerespulmonares de células pequeñas, y la secreción ectópica de HC a partir de tumores no endocrinos.
Manifestaciones clínicas
El trastorno suele tener inicio insidioso y los síntomas muchas veces persisten durante un período considerable antes de que se establezca el diagnóstico. Cuando la síntesis de HC excesiva ocurre una vez que las epífisis de los huesos largos se cierran, como en el adulto, la persona ya no puede aumentar de talla, pero los tejidos blandos siguen creciendo. El crecimiento de los huesos pequeños de las manos y pies y de los huesos membranosos de la cara y el cráneo da origen al crecimiento pronunciado de las manos y los pies, al desarrollo de una nariz ancha y bulbosa, a la protrusión de la mandíbula y al aplanamiento de la frente (figura 49-2).
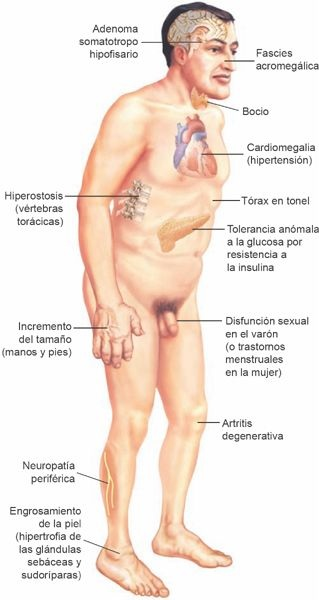
Los dientes se separan, lo que provoca anomalías de la mordida y dificultad para la masticación. Las estructuras cartilaginosas de la laringe y las vías respiratorias también crecen, lo que da origen a un engrosamiento de la voz y a la tendencia al desarrollo de bronquitis. Los cambios vertebrales muchas veces determinan cifosis o jorobamiento. El sobrecrecimiento de los huesos conduce con frecuencia al desarrollo de artralgias y artritis degenerativa de la columna, las caderas y las rodillas. Casi todos los órganos del organismo aumentan de tamaño. El crecimiento cardíaco y la ateroesclerosis acelerada pueden inducir una muerte súbita.
Los efectos metabólicos de las concentraciones excesivas de HC incluyen alteraciones del metabolismo de los lípidos y carbohidratos. La HC induce aumento de la liberación de ácidos grasoslibres a partir del tejido adiposo, lo que determina una mayor concentración de estas sustancias en los fluidos corporales. Además, la HC aumenta la formación de cetonas y el consumo de ácidos grasos libres para la obtención de energía, de manera preferencial al empleo de carbohidratos y proteínas.
La HC ejerce efectos múltiples sobre el metabolismo de los carbohidratos, como la disminución de la captación de la glucosa en los tejidos como el músculo esquelético y el tejido adiposo, el aumento de la producción de glucosa en el hígado, y el aumento de la secreción de insulina. Cada uno de estos cambios da origen a la resistencia a la insulina inducida por HC. Esto conduce a la intolerancia a la glucosa, que estimula a las células β del páncreas para sintetizar insulina adicional. La elevación a largo plazo de la HC tiene como consecuencia la estimulación excesiva de las células β y las hace «quemarse» literalmente. Las anomalías de la tolerancia a la glucosa se presentan hasta en el 50% al 70% de los individuos con acromegalia. De manera subsecuente puede desarrollarse diabetes mellitus franca.
La glándula hipófisis se ubica en la fosa hipofisaria del hueso esfenoides (es decir, la silla turca), que se ubica justo por debajo del quiasma óptico. El crecimiento de la glándula hipófisis induce erosión del hueso circundante y, por efecto de su ubicación, esto puede desencadenar cefalea, defectos del campo visual que derivan de la compresión del quiasma óptico (de manera clásica, hemianopsia bitemporal) y parálisis de los nervios craneales III, IV y VI. La compresión del otras estructuras hipofisarias puede inducir hipotiroidismo secundario, hipogonadismo e insuficiencia suprarrenal. El hipogonadismo puede ser consecuencia de un daño directo al sistema hipotalámico o hipofisario, o derivar de manera indirecta de la hiperprolactinemia por la incapacidad del factor inhibidor de la prolactina (dopamina) para alcanzar a los lactotropos hipofisarios (células que secretan prolactina) por el daño generado por el tumor hipofisario.
Otras manifestaciones incluyen la diaforesis excesiva con olor desagradable, la piel grasosa, la intolerancia al calor, la ganancia moderada de peso, la debilidad muscular y la fatiga, las irregularidades menstruales, y la disminución de la libido. La hipertensión es más bien común. El síndrome de apnea del sueño se identifica en el hasta 90% de los enfermos. La patogénesis del síndrome de apnea del sueño es la obstrucción en la mayor parte de los individuos por efecto del aumento de la acumulación de tejidos blandos faríngeos. Pueden desarrollarse parestesias como resultado del atrapamiento y la compresión de los nervios por los tejidos blandos excesivos y la acumulación de líquido subcutáneo (en particular, el síndrome del túnel del carpo). La acromegalia también se relaciona con un aumento del riesgo de desarrollo de pólipos colónicos y cáncer colorrectal. La tasa de mortalidad en las personas con acromegalia es de 2 a 3 veces la esperada, en su mayoría por trastornos cardiovasculares y cáncer. La enfermedad cardiovascular deriva de una combinación de miocardiopatía, hipertensión, resistencia la insulina, e hiperinsulinemia e hiperlipidemia.
Diagnóstico
La acromegalia se desarrolla con frecuencia de manera insidiosa y sólo un número escaso de personas busca atención médica por efecto de los cambios en su aspecto. El diagnóstico de la acromegalia se facilita al reconocer las características típicas del trastorno: crecimiento de las manos y los pies, y desarrollo de rasgos faciales toscos. Las pruebas de laboratorio para la detección de concentraciones elevadas de HC que no se suprimen con una carga de glucosa se utilizan para confirmar el diagnóstico. Los estudios de IRM pueden detectar y localizar las lesiones hipofisarias. Puesto que la mayor parte de los efectos de la HC se encuentra mediada por el FCI-1, sus concentraciones pueden arrojar información en torno a la actividad del trastorno.
Tratamiento
El tratamiento de la acromegalia se concentra en la corrección de las anomalías metabólicas, la normalización de las concentraciones del FCI-1 hasta alcanzar los niveles de loscontroles correspondientes para la edad y el sexo, el retiro o la reducción de la masa tumoral, el alivio de los efectos de compresión central y el mejoramiento de las características clínicas adversas. Los tumores hipofisarios pueden extirparse por medios quirúrgicos mediante una técnica transesfenoidal. El tratamiento médico suele proporcionarse con un papel adyuvante. Los análogos de la somatostatina inducen una inhibición por retroalimentación de la HC, y son efectivos para el manejo médico de la acromegalia. Los agonistas de la dopamina reducen las concentraciones de HC y se han utilizado para el manejo médico de la acromegalia. Los antagonistas de los receptores de HC son análogos de la HC humana en los que se Modifican la estructura. Los antagonistas del receptor de la HC se unen a estas estructuras en todas las superficies celulares, donde bloquean la unión de la HC endógena e interfieren así con la transducción de señales inducida por HC.
Pubertad precoz isosexual
La pubertad precoz isosexual se define como una activación temprana del eje hipotálamo-hipófiso-gonadal, que da origen al desarrollo de características sexuales apropiadas y fertilidad. La pubertad precoz se define ahora como la aparición de desarrollo sexual secundario antes de la edad de 7 años en niñas caucásicas y de 6 años en niñas afroamericanas. En los niños de ambas etnias el límite inferior de la edad aún es de 9 años. Sin embargo, se reconoce que la pubertad puede desarrollarse en forma más temprana en los varones con obesidad. El desarrollo sexual precoz puede ser idiopático o derivar de la enfermedad gonadal, suprarrenal o hipotalámica. Los tumores benignos y malignos del sistema nervioso central (SNC) pueden inducir pubertad precoz. Se piensa que estos tumores pueden eliminar las influencias inhibidoras que normalmente se ejercen sobre el hipotálamo durante la niñez.
El diagnóstico de la pubertad precoz se basa en los hallazgos físicos de telarca (es decir, inicio del desarrollo mamario), adrenarca (es decir, inicio del aumento de la síntesis de andrógenos suprarrenales) y menarca (es decir, inicio de la función menstrual) tempranas en las niñas. El signo más frecuente en los varones es el crecimiento genital temprano. Los hallazgos radiológicos pueden revelar edad ósea avanzada. Las personas con pubertad precoz suelen ser altas para su edad durante la niñez, pero tener peso bajo durante la edad adulta, por efecto del cierre temprano de las epífisis. Deben utilizarse IRM o tomografía computarizada (TC) para excluir lesiones intracraneales.
Según la etiología de la pubertad precoz, pudiera no necesitarse tratamiento o requerirse cirugía, o consumo de fármacos. La administración de un agonista de HLGn de acción prolongada permite una disminución de la capacidad de respuesta hipofisaria a la HLGn, lo que da lugar a una disminución de la secreción de hormonas gonadotrópicas y esteroides sexuales (es decir, por una regulación negativa de los receptores de HLGn).