06. Enfermedad cardíaca en lactantes y niños
Aunque cada año miles de lactantes tendrá una cardiopatía congénita, otros niños desarrollarán un trastorno cardíaco adquirido, incluida la enfermedad de Kawasaki.
Desarrollo embrionario del corazón
El corazón es el primer órgano funcional del embrión. Sus primeros movimientos pulsátiles comienzan en la tercera semana después de la concepción. Este desarrollo temprano del corazón es esencial para el embrión que crece con rapidez como una forma de circular los nutrimentos y eliminar los productos de desecho. La mayor parte del desarrollo del corazón y los vasos sanguíneos ocurre entre la tercera y la octava semanas de vida embrionaria.
El corazón en desarrollo comienza como 2 tubos de endotelio que se fusionan en una sola estructura tubular. Las estructuras cardíacas tempranas se desarrollan conforme el corazón se alarga y adquiere dilataciones y constricciones alternadas. Primero se desarrollan una sola aurícula y un ventrículo, junto con el bulbo del corazón (figura 32-21). A esto le sigue la formación del tronco arterioso y el seno venoso, un espacio venoso grande que recibe sangre del embrión y la placenta en desarrollo. Los movimientos pulsátiles tempranos del corazón comienzan en el seno venoso y desplazan la sangre fuera del corazón a través del bulbo del corazón, el tronco arterioso y los arcos aórticos.
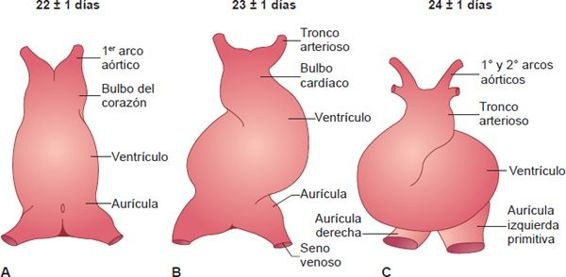
El ritmo de crecimiento distinto de las estructuras cardíacas primitivas, junto con la fijación del corazón en sus extremos venoso y arterial, hacen que el corazón tubular se flexione sobre sí mismo.
Conforme el corazón se dobla, la aurícula y el seno venoso quedan detrás del bulbo cardíaco, el tronco arterioso y el ventrículo. Este plegamiento del corazón primitivo da lugar a la alineación del corazón en el lado izquierdo del tórax, con la aurícula situada detrás del ventrículo. La rotación anómala durante la formación del asa ventricular puede causar varias posiciones anómalas, como la dextroposición del corazón.
El corazón embrionario continúa su desarrollo con la división de las cámaras. El fraccionamiento del conducto AV, la aurícula y el ventrículo comienza en la cuarta semana, para la quinta está completo. La tabicación del corazón comienza cuando se forman paquetes tisulares llamados cojinetes endocárdicos en la parte media de las paredes dorsal y ventral del corazón, en la región del conducto AV, y comienzan a crecer hacia el centro. Hasta que comienza la tabicación, existe un solo conducto AV ente las aurículas y los ventrículos. Conforme crecen los cojinetes endocárdicos, se unen y fusionan para separar los conductos AV derecho e izquierdo (figura 32-22).
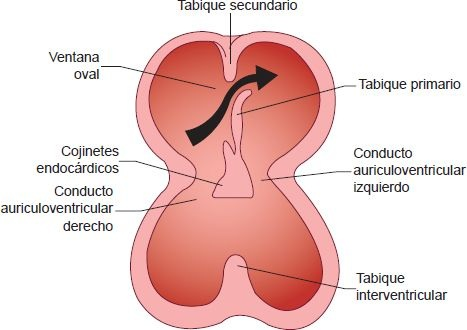
Las válvulas mitral y tricúspide se desarrollan en estos conductos. Los cojinetes endocárdicos también contribuyen a la formación de partes de los tabiques auricular y ventricular. Los defectos en la formación del cojinete endocárdico pueden causar anomalías septales auriculares y ventriculares, defectos completos en el conducto AV y anomalías en las válvulas mitral y tricúspide.
La separación de los ventrículos comienza con el crecimiento del tabique interventricular desde el piso del ventrículo hacia arriba, a los cojinetes endocárdicos. La fusión de los cojinetes endocárdicos con el tabique interventricular se completa hacia el final de la séptima semana.
La división del tabique auricular es más compleja y ocurre en 2 etapas; comienza con la formación de una delgada membrana en forma de medialuna llamada tabique primario que surge de la parte anterosuperior del corazón y crece hacia los cojinetes endocárdicos, pero deja una abertura llamada orificio primario (foramen primum) entre su borde inferior y los cojinetes endocárdicos. Una segunda membrana, llamada tabique secundario, también comienza a crecer de la pared superior de la aurícula, a la derecha del tabique primario. Conforme esta membrana crece hacia los cojinetes endocárdicos, se superpone poco a poco sobre una abertura en la parte superior del tabique primario, con lo que se forma una abertura oval con una válvula del tipo solapa llamada ventana oval (figura 32-22). La parte superior del tabique primitivo desaparece de manera gradual, la parte restante se convierte en la válvula de la ventana oval. La ventana oval constituye una vía de comunicación entre las 2 cámaras superiores del corazón. Esta abertura, que casi siempre se cierra poco después del nacimiento, permite que la sangre de la vena umbilical llegue de manera directa al lado izquierdo del corazón, sin pasar por los pulmones.
Para completar la transformación en un corazón de 4 cámaras, debe haber mecanismos que separen la sangre bombeada del lado derecho del corazón, que se desvía a la circulación pulmonar,de la sangre bombeada por el lado izquierdo del corazón, que se dirige a la circulación sistémica.
Esta separación del flujo se acompaña de cambios en el desarrollo de los conductos de salida del corazón tubular, el bulbo del corazón y el tronco arterioso (figura 32-23). Conforme estos vasos adquieren una forma espiral y se dividen, la aorta toma su posición posterior y a la derecha de la arteria pulmonar. La formación alterada de espirales durante esta etapa del desarrollo puede generar defectos como la trasposición de los grandes vasos.
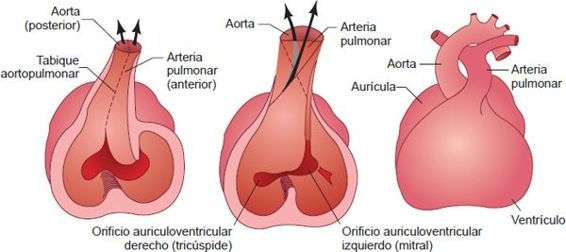
En el proceso de formar un tronco pulmonar y aorta separados, se desarrolla un vaso llamado conducto arterioso. Este vaso, que conecta la arteria pulmonar y la aorta, permite que la sangre que entra al tronco pulmonar se desvíe hacia la aorta como una forma de evitar el paso por los pulmones.
Como la ventana oval, el conducto arterioso casi siempre se cierra poco después del nacimiento.
Circulación fetal y perinatal
La circulación fetal tiene diferencias anatómicas y fisiológicas con respecto a la circulación posnatal.
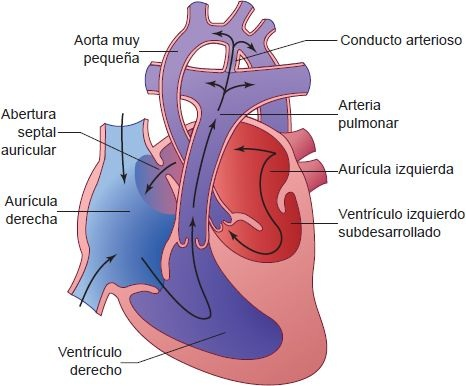
El flujo sanguíneo en la circulación fetal es paralelo, no en serie, la mayor parte del gasto del ventrículo derecho se dirige a la placenta para captación de oxígeno y el VI bombea sangre al corazón, cerebro y a la parte superior del feto, sobre todo. Antes del nacimiento, la oxigenación sanguínea ocurre en la placenta, después del nacimiento ocurre en los pulmones. El feto se mantiene en un estado con baja oxigenación (PO2 30 mm Hg a 35 mm Hg; saturación de hemoglobina con O2, del 60% al 70%). Para compensarlo, el gasto cardíaco fetal es más alto que en cualquier otra etapa de la vida (400 ml/kg/min a 500 ml/kg/min) y la hemoglobina fetal tiene una mayor afinidad por el oxígeno. Además, los vasos pulmonares del feto están muy constreñidos porque los pulmones están llenos de líquido y porque en el feto existe un estímulo hipóxico intenso para la vasoconstricción.
Como resultado, el flujo sanguíneo por los pulmones es menor que en cualquier otro momento de la vida.
En el feto, la sangre entra a la circulación por la vena umbilical y regresa a la placenta por las 2 arterias umbilicales (figura 32-24).
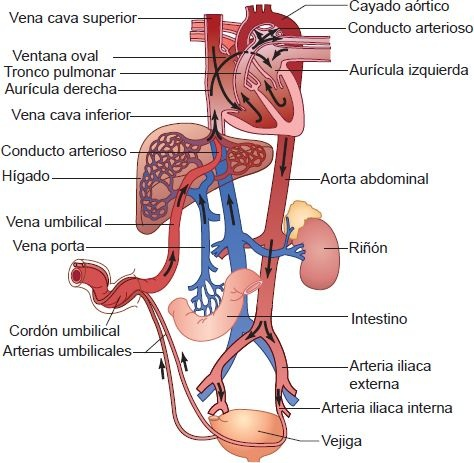
Un vaso llamado conducto venoso permite que la mayor parte de la sangre de la vena umbilical evite el paso por la circulación hepática y llegue directamente a la vena cava inferior. La sangre fluye de la vena cava inferior a la aurícula derecha, donde cerca del 40% del volumen sanguíneo pasa por la ventana oval a la aurícula izquierda, luego al ventrículo izquierdo y se expulsa a la aorta ascendente para irrigar la cabeza y las extremidades superiores. De esta manera, la sangre mejor oxigenada de la placenta se utiliza para la perfusión cerebral. Al mismo tiempo, la sangre venosa de la cabeza y las extremidades superiores regresa al lado derecho del corazón a través de la vena cava superior, pasa al ventrículo derecho y se expulsa por la arteria pulmonar.
Debido a la resistencia vascular pulmonar muy alta, casi el 90% de la sangre expulsada a la arteria pulmonar se desvía por el conducto arterioso a la aorta descendente. Esta sangre irriga las extremidades inferiores y regresa a la placenta por las arterias umbilicales.
Al nacer, el lactante respira por primera vez y cambia de la oxigenación placentaria de la sangre a la oxigenación pulmonar. Los cambios más drásticos en la circulación después del nacimiento son la eliminación del lecho vascular placentario de baja resistencia y la vasodilatación pulmonar marcada que se produce con el inicio de la ventilación. Unos minutos después del nacimiento, el flujo sanguíneo pulmonar aumenta de 35 ml/kg/min a 160 ml/kg/min O200 ml/kg/min. La presión en la circulación pulmonar y el lado derecho del corazón cae conforme el líquido pulmonar se sustituye por aire y conforme la expansión pulmonar disminuye la presión transmitida a los vasos sanguíneos pulmonares. Con la inflación pulmonar, la tensión alveolar de oxígeno aumenta, lo que revierte la vasoconstricción pulmonar inducida por la hipoxemia en la circulación fetal.
El pinzamiento del cordón y la eliminación de la circulación placentaria de baja resistencia producen un aumento en la resistencia vascular sistémica, con un aumento consecuente en la presión ventricular izquierda. El descenso resultante en la presión auricular derecha y el incremento en la presión auricular izquierda producen el cierre de la válvula de tapa de la ventana oval. La reversión del estado hipoxémico fetal también causa constricción del músculo liso ductal, lo que contribuye al cierre del conducto arterioso a las 72 h después del nacimiento. Luego del descenso súbito inicial de la resistencia vascular pulmonar, existe un descenso más gradual en la resistencia vascular pulmonar derivado de la regresión de la capa muscular lisa medial en las arterias pulmonares. Durante las primeras 2 a 9 semanas de edad, el adelgazamiento gradual de la capa de músculo liso produce descensos adicionales en la resistencia vascular pulmonar. Para cuando un lactante a término saludable tiene varias semanas de edad, la resistencia vascular pulmonar ya descendió a los niveles del adulto.
El desarrollo vascular pulmonar posnatal puede alterarse por varios factores, que incluyen hipoxia alveolar, premadurez, enfermedad pulmonar y defectos cardíacos congénitos. La hipoxia alveolar es uno de los estímulos más potentes para la vasoconstricción pulmonar y la hipertensión pulmonar en el recién nacido. Durante este período, las arterias pulmonares se mantienen muy reactivas y pueden constreñirse como respuesta a la hipoxia, acidosis, hiperinflación alveolar e hipotermia. Por tanto, la hipoxia durante los primeros días de edad puede retrasar o impedir el descenso normal en la resistencia vascular pulmonar.
Gran parte del desarrollo de la capa muscular lisa en las arteriolas pulmonares ocurre durante la gestación; como resultado, los lactantes prematuros tienen menos músculo liso en la túnica media. Estos lactantes siguen el mismo patrón de regresión del músculo liso, pero como existe menor cantidad de músculo, la capa muscular regresa en menos tiempo. El músculo liso vascular pulmonar de los lactantes prematuros también tiene menor capacidad de respuesta a la hipoxia. Por estas razones, un lactante prematuro puede tener un descenso más marcado en la resistencia vascularpulmonar, con la desviación consecuente de sangre de la aorta por el conducto arterioso a la arteria pulmonar unas horas después de nacer.
Defectos cardíacos congénitos
El desarrollo principal del corazón fetal ocurre entra la cuarta y la séptima semanas de gestación, y la mayoría de los defectos cardíacos congénitos se producen en ese período. Se cree que la mayoría de los defectos cardíacos congénitos son de origen multifactorial, se deben a la interacción de la predisposición genética al desarrollo de estas anomalías y las influencias ambientales.
El conocimiento de la base genética de los defectos cardíacos congénitos ha aumentado mucho en los últimos años. Esta área de investigación adquiere una importancia particular, ya que más personas con cardiopatías congénitas sobreviven hasta la edad adulta y consideran tener hijos. El conocimiento reciente sugiere que la contribución genética a la cardiopatía congénita se había subestimado hasta ahora. Algunos defectos cardíacos, como la estenosis aórtica, defecto en el tabique interauricular del tipo secundario, estenosis valvular pulmonar, tetralogía de Fallot y ciertos defectos en el tabique interventricular, tienen una predisposición familiar más marcada que otros.
Los defectos cardíacos congénitos también se han vinculado con anomalías cromosómicas, hasta el 30% de los niños con cardiopatía congénita tiene alguna anomalía cromosómica. Existe enfermedad cardíaca casi en el 100% de los niños con trisomía 18, en el 50% de los que tienen trisomía y en el 35% de aquellos con síndrome de Turner. Otro síndrome que a menudo incluye malformaciones cardíacas es el de Williams (microdelección 7q11,23), que se relaciona con estenosis aórtica supravalvular y estenosis pulmonar.
Hasta el 30% de los defectos cardíacos congénitos pueden atribuirse a factores de riesgo identificables y modificables, como influencias teratógenas y trastornos maternos como enfermedades febriles, lupus eritematoso sistémico, diabetes mellitus, consumo materno de alcohol y tratamiento con anticonvulsivos, retinoides, litio y otros fármacos con o sin prescripción. La atención prenatal adecuada, sobre todo la ingestión de multivitamínico con ácido fólico antes y después de la concepción, puede reducir el riesgo de enfermedad cardíaca en el feto.
Fisiopatología
Los principales efectos de los defectos cardíacos congénitos ocurren por la desviación anómala de la sangre, generación de cianosis y alteración del flujo sanguíneo pulmonar.
Desviación anómala de la sangre
La desviación de la sangre se refiere al cambio del flujo sanguíneo de un sistema a otro: del sistema arterial al venoso (cortocircuito de izquierda a derecha) o del sistema venoso al arterial (cortocircuito de derecha a izquierda). La desviación de sangre en los defectos cardíacos congénitos depende de la presencia, posición y tamaño de una abertura anómala entre las circulaciones derecha e izquierda, y del grado de resistencia al flujo a través de la abertura.
La resistencia vascular de las circulaciones sistémica y pulmonar influye en la dirección del cortocircuito. Debido a la elevada resistencia vascular pulmonar en el recién nacido, los defectos en el tabique interauricular e interventricular casi nunca producen una desviación significativa o síntomas durante las primeras semanas de edad.
Conforme el músculo liso vascular pulmonar regresa en el recién nacido, la resistencia de la circulación pulmonar cae por debajo de la resistencia en la circulación sistémica; en los defectos septales auriculares o ventriculares no complicados, la sangre se desvía del lado izquierdo del corazón al derecho. En los defectos más complicados del tabique interventricular, la resistencia alta al flujo de salida puede afectar el patrón de la derivación. Por ejemplo, los defectos que aumentan la resistencia al flujo de salida aórtico (ej. estenosis valvular aórtica, coartación aórtica, síndrome de hipoplasia cardíaca izquierda) aumentan el cortocircuito de izquierda a derecha, y los defectos que obstruyen el flujo de salida pulmonar (ej. estenosis valvular pulmonar, tetralogía de Fallot) aumentan el cortocircuito de derecha a izquierda. El llanto, la defecación e incluso el estrés de la alimentación pueden aumentar la resistencia vascular pulmonar e incrementar el cortocircuito de derecha a izquierda y la cianosis en lactantes con defectos septales.
Trastornos cianóticos y no cianóticos
Las cardiopatías congénitas a menudo se dividen en 2 categorías: no cianóticas y cianóticas. Los defectos que producen un cortocircuito de izquierda a derecha casi siempre se clasifican como no cianóticos porque no afectan la oxigenación sanguínea en la circulación pulmonar.
Los defectos que desvían la sangre del lado derecho del corazón al izquierdo o que obstruyen el flujo sanguíneo pulmonar se clasifican como trastornos cianóticos. La cianosis, una coloración azulada de la piel, más notable en los lechos ungueales y las mucosas, aparece cuando suficiente sangre desoxigenada del lado derecho del corazón se mezcla con sangre oxigenada del lado izquierdo del corazón. El color anómalo se vuelve evidente cuando la saturación de oxígeno es menor del 80% en los capilares (equivale a 5 g de hemoglobina desoxigenada).
Un cortocircuito de derecha a izquierda hace que la sangre desoxigenada del lado derecho del corazón pase al lado izquierdo y luego se expulse a la circulación sistémica. Con un cortocircuito de izquierda a derecha, la sangre oxigenada que debía expulsarse a la circulación sistémica regresa al lado derecho del corazón y de nuevo a los pulmones. Este aumento del volumen distiende el lado derecho del corazón y la circulación pulmonar, incrementa la carga de trabajo del ventrículo derecho. Un niño con un defecto que produce un cortocircuito de izquierda a derecha casi siempre tiene crecimiento del lado derecho del corazón y los vasos sanguíneos pulmonares.
De los defectos congénitos descritos en este capítulo, la persistencia del conducto arterioso, defectos septales auriculares y ventriculares, defectos en el cojinete endocárdico, estenosis valvular pulmonar y coartación aórtica se consideran defectos con cianosis leve o nula; la tetralogía de Fallot, la trasposición de grandes vasos y la anatomía con ventrículo único son defectos con cianosis.
Alteración del flujo sanguíneo pulmonar
Muchas de las complicaciones de las cardiopatías congénitas se deben al descenso o aumento en el flujo sanguíneo pulmonar. Los defectos que reducen el flujo sanguíneo pulmonar (ej. estenosis pulmonar) casi siempre causan fatiga, disnea y falta de progreso. En contraste con las arteriolas de la circulación sistémica, las de la circulación pulmonar son vasos de paredes delgadas que se adaptan a diversos niveles de volumen por latido expulsado del ventrículo derecho.
El adelgazamiento de los vasos pulmonares ocurre durante las primeras semanas después del nacimiento, durante las cuales la media de los vasos se adelgaza y la resistencia vascular pulmonar disminuye. En un lactante a término con un defecto cardíaco congénito que incrementa mucho el flujo sanguíneo pulmonar (ej. defecto septal ventricular), el flujo aumentado estimula la vasoconstricción pulmonar y retrasa o reduce el adelgazamiento involutivo normal de las pequeñas arteriolas pulmonares. En la mayoría de los casos, durante la lactancia temprana la resistencia vascular sólo aumenta un poco y la contribución principal a la hipertensión pulmonar es el aumento en el flujo sanguíneo. Sin embargo, en algunos lactantes con un cortocircuito grande de derecha a izquierda, la resistencia vascular pulmonar nunca disminuye.
Los defectos cardíacos congénitos que producen una elevación persistente del flujo sanguíneo pulmonar o la resistencia vascular pulmonar tienen la capacidad de causar hipertensión pulmonar y cambios patológicos irreversibles en los vasos sanguíneos pulmonares. Cuando la desviación del flujo sanguíneo sistémico hacia la circulación pulmonar puede causar lesión permanente a los vasos pulmonares, debe practicarse un procedimiento quirúrgico para reducir el flujo de manera temporal o permanente. La colocación de bandas en la arteria pulmonar consiste en aplicar una banda constrictiva alrededor de la arteria pulmonar principal, lo que aumenta la resistencia al flujo de salida del ventrículo derecho. Esta técnica es una medida temporal para aliviar síntomas y proteger los vasos sanguíneos pulmonares como anticipación a una reparación quirúrgica ulterior del defecto.
Manifestaciones y tratamiento
Cada vez es más frecuente el diagnóstico prenatal de los defectos cardíacos. En este caso, el lactante puede evaluarse poco después del nacimiento para confirmar el diagnóstico y desarrollar un plan terapéutico. Pueden obtenerse imágenes diagnósticas confiables del corazón fetal desde las 16 semanas de gestación, y existen estudios en los que se emplea la ecografía transvaginal para visualizar el corazón incluso antes de eso. Entre los trastornos que pueden diagnosticarse con certeza mediante ecocardiografía fetal están los defectos en el tabique AV, síndrome de hipoplasia cardíaca izquierda, estenosis valvular aórtica, miocardiopatía hipertrófica, estenosis valvular pulmonar y trasposición de grandes arterias. Es más probable que se detecten los trastornos que producen alguna alteración en la vista de 4 cámaras, una imagen típica durante la ecografía prenatal de rutina.
En el período posnatal, los defectos cardíacos congénitos pueden manifestarse con muchos signos y síntomas. Se han descrito más de 40 tipos distintos de defectos cardíacos congénitos e incluso las lesiones individuales tienen espectros amplios de gravedad. Por lo tanto, no existe una forma estándar de manifestación entre los lactantes y niños con cardiopatía congénita. Algunos defectos, como la persistencia del conducto arterioso y los defectos pequeños en el tabique interventricular, se cierran de manera espontánea. Otros defectos menos graves a veces no causan manifestaciones evidentes y el trastorno se detecta en un examen médico de rutina. La cianosis, congestión pulmonar, insuficiencia cardíaca y la hipoperfusión periférica son las principales preocupaciones en niños con defectos más graves. Estos defectos a menudo causan problemas desde el nacimiento o en la lactancia temprana. Es posible que el niño tenga cianosis, dificultad respiratoria y fatiga fácil, y es probable que tenga dificultad para alimentarse y falta de progreso. La cianosis generalizada que persiste más de 3 h después del nacimiento sugiere cardiopatía congénita.
Una prueba de exposición a oxígeno (administración de oxígeno al 100% por 10 min) ayuda a determinar si existe cardiopatía congénita en un recién nacido cianótico. Durante el período de exposición se obtiene una muestra de sangre arterial. Si la presión parcial de oxígeno (PO2) es mayor de 250 mm Hg, puede descartarse la cardiopatía cianótica; si la PO2 está entre 160 mm Hg y 250 mm Hg, la cardiopatía es improbable; la falta de aumento en la PO2 hasta estas cifras es muy sugestiva de cardiopatía cianótica. Como la cianosis infantil puede verse como coloración oscura de la piel, es importante valorar el color de las mucosas, las uñas de las manos y pies, la lengua y los labios. La congestión pulmonar en un lactante produce aumento de la frecuencia respiratoria, ortopnea, gruñidos, sibilancias, tos y estertores. Una radiografía torácica permite distinguir con rapidez entre los lactantes con disminución de las marcas vasculares pulmonares (densidades) de los que tienen marcas normales o aumentadas. El lactante con disminución marcada de la perfusión periférica puede estar en estado de shock.
La insuficiencia cardíaca se manifiesta como taquipnea o disnea en reposo o con el esfuerzo. Para el lactante, esto suele suceder durante la alimentación. También puede haber infecciones respiratorias recurrentes y transpiración excesiva. Además puede haber síncope o casi síncope. La falta de crecimiento se debe a la insuficiencia cardíaca no resuelta. El plan terapéutico casi siempre incluye medidas de apoyo (ej. digoxina, diuréticos y complementación alimentaria) diseñadas para ayudar al lactante a compensar las limitaciones en la reserva cardíaca y a prevenir las complicaciones. A menudo es necesaria la intervención quirúrgica para los defectos graves; puede hacerse en las primeras semanas de edad o, si las condiciones lo permiten, se pospone hasta que el niño sea mayor. Los niños con cardiopatía congénita estructural y los que se sometieron a cirugía correctiva tienen un riesgo de EI mayor al anticipado.
Se sugiere el tratamiento antibiótico profiláctico antes de procedimientos dentales u otros períodos de riesgo elevado de bacteriemia para niños con:
- Cardiopatía cianótica no reparada, incluidos los que tienen cortocircuitos y conductos paliativos.
- Cardiopatía congénita reparada por completo con material o un dispositivo prostéticos, ya sea que se situén en una intervención quirúrgica o por catéter, durante los 6 meses siguientes al procedimiento.
- Cardiopatía congénita reparada con defectos residuales en el sitio o junto al sitio de un parche, o un dispositivo prostético (que inhibe la endotelización) y antes de IE.
Tipos de defectos
Los defectos cardíacos congénitos pueden afectar casi cualquier estructura del corazón o de los vasos sanguíneos centrales. Los defectos incluyen comunicación entre las cámaras cardíacas; desarrollo interrumpido de las cámaras cardíacas o estructuras valvulares; posición anómala de las cámaras cardíacas y grandes vasos; y cierre anómalo de los conductos fetales de comunicación. El defecto particular refleja la etapa del desarrollo embrionario en el que ocurrió. Es frecuente que haya defectos múltiples en un niño y en el caso de algunos defectos, como la tetralogía de Fallot, que incluyan varias anomalías.
El desarrollo del corazón es simultáneo y secuencial; un defecto cardíaco puede reflejar los múltiples episodios del desarrollo que ocurrían al mismo tiempo o en secuencia. La mayoría de los lactantes con cardiopatía congénita no tiene problemas mayores durante la lactancia. Sólo cerca de un tercio de los lactantes con anomalías tiene una enfermedad crítica. Se han identificado más de 40 tipos de defectos, los más frecuentes son los que afectan el tabique interventricular (del 28% al 42%).
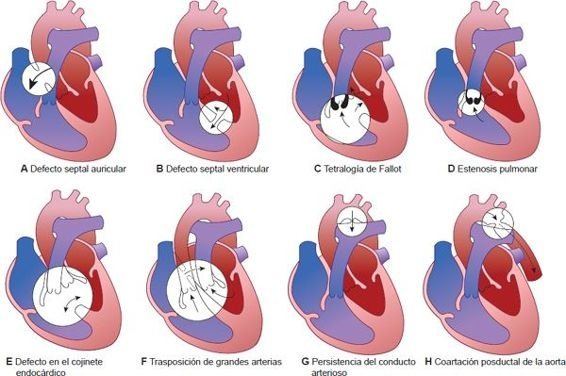
Persistencia del conducto arterioso
El conducto arterioso tiene un papel vital para desviar sangre del lado derecho del corazón y los pulmones hacia la circulación sistémica durante la etapa fetal (figura 32-25G). Con el inicio de la respiración espontánea después del nacimiento, este vaso casi siempre se cierra por constricción muscular del tejido ductal. Se cree que el primer paso del cierre ductal en el lactante sano es el aumento súbito en la saturación arterial de oxígeno y la caída subsiguiente de la resistencia vascular pulmonar después del nacimiento.
Los factores adicionales considerados contribuyentes al cierre ductal son la caída en la concentración de prostaglandinas y adenosinas, y la liberación de sustancias vasoactivas. Después de la constricción, el lumen del conducto se sella de manera permanente con tejido fibroso en 2 o 3 semanas.
Para el 90% de los lactantes a término, el conducto se mantiene funcionalmente cerrado a las 48 h de edad. Es más probable que el conducto arterioso se mantenga permeable en los lactantes a término con anomalías circulatorias o ventilatorias y en los lactantes prematuros. La oxigenación arterial, las prostaglandinas circulantes, los factores genéticos y otros factores desconocidos interactúan para determinar el mecanismo de cierre ductal. La concentración de prostaglandina circulante tiene relación directa con la edad gestacional y la incidencia de persistencia del conducto arterioso en lactantes que pesan menos de 1.000 g al nacer puede ser de hasta el 50%.
La permeabilidad persistente del conducto arterioso se define como el que permanece abierto después de 3 meses en un lactante a término. El tamaño del conducto permeable y la diferencia entre la resistencia vascular sistémica y la pulmonar determinan las manifestaciones clínicas. Por lo general, la sangre se desvía por el conducto del lado izquierdo de presión más alta (circulación sistémica) al lado derecho de presión menor (circulación pulmonar).
Por lo general, se detecta un soplo días o semanas después del nacimiento. El soplo es más intenso en el segundo espacio intercostal izquierdo, es continuo durante la sístole y la diástole, y tiene un sonido característico de «maquinaria». A menudo, la presión ampliada del pulso se debe al flujo continuo de sangre aórtica hacia la arteria pulmonar. Los métodos diagnósticos incluyen radiografía torácica y ecocardiografía.
En la radiografía torácica se observa aumento en las marcas pulmonares y crecimiento del lado izquierdo del corazón por el incremento del retorno venoso si el cortocircuito es grande. Las radiografías torácicas son normales si el cortocircuito es pequeño. La ecocardiografía se emplea para determinar la presencia, tamaño, dirección (o sea, de izquierda a derecha o de derecha a izquierda) y las consecuencias físicas del cortocircuito.
Un conducto permeable no tratado puede causar complicaciones sustanciales en el largo plazo que incluyen insuficiencia cardíaca congestiva, EI, enfermedad vascular pulmonar, formación de aneurisma, tromboembolia y calcificación. La posibilidad de complicaciones y la morbilidad y mortalidad tan bajas del procedimiento justifican el cierre del conducto arterioso permeable, incluso cuando el cortocircuito sea pequeño.
En el lactante prematuro, el conducto permeable puede causar dificultad respiratoria e impedir la separación del ventilador mecánica. La indometacina, un inhibidor de la síntesis de prostaglandina, es efectiva hasta en el 79% de los lactantes prematuros. También se ha obtenido cierto éxito con el consumo de ibuprofeno, pero todavía se desconocen los efectos de largo plazo en la enfermedad pulmonar crónica y la hipertensión pulmonar.
Cuando este tratamiento médico falla, se recomienda la intervención quirúrgica. En el lactante a término o el niño mayor, el cierre puede hacerse por ligadura quirúrgica u oclusión con un dispositivo. Por lo general, la cirugía implica una toracotomía izquierda pequeña o toracoscopia que permita ligar el vaso. Los dispositivos implantables, casi siempre espirales, permiten el cierre exitoso del conducto en el laboratorio de cateterismo como procedimiento ambulatorio. La anatomía del conducto y el tamaño del paciente son los factores clave para decidir si esta técnica es aplicable.
Aunque siempre se recomienda el cierre del conducto arterioso cuando existe como lesión aislada, el mantenimiento deliberado de la permeabilidad del conducto puede ser una medida salvadora de la vida en niños con formas complejas de cardiopatía congénita que tienen flujo sanguíneo pulmonar o sistémico dependiente del conducto, o en pacientes con mezcla obligada de las circulaciones arterial y venosa (trasposición de grandes arterias). La infusión intravenosa de prostaglandina E1 ha sido muy efectiva para mantener la permeabilidad del conducto o reabrir el conducto en los recién nacidos. En la actualidad, este tratamiento se emplea siempre en los recién nacidos con sospecha de defectos cardíacos congénitos hasta que puedan trasladarse a un centro especializado donde se confirme un diagnóstico.
Defectos en el tabique interauricular
Cualquier abertura persistente que permita la desviación de la sangre a través del tabique interauricular se considera un defecto septal auricular. La anomalía puede ser única o múltiple y varía desde una pequeña abertura asintomática hasta una grande causante de síntomas. La tipología del defecto depende de su posición y puede incluir un defecto auricular secundario (la forma más frecuente), un defecto en el orificio primario, un defecto en el seno venoso o persistencia de la ventana oval (figura 32-25A). La anomalía es más frecuente en las niñas que en los varones, con una proporción de hasta 2:1. Hasta el 50% de los niños con cardiopatía congénita tienen un defecto septal auricular como parte del diagnóstico.Muchos defectos en el tabique interauricular son asintomáticos y se descubren durante un examen médico de rutina a los pocos años de edad.
El cortocircuito intracardíaco casi siempre es de izquierda a derecha y puede aumentar con la edad, conforme aumenta la distensibilidad del ventrículo derecho. En la mayoría de los casos existe un cortocircuito moderado que causa dilatación de las cámaras cardíacas derechas y perfusión excesiva de la circulación pulmonar. El aumento en el volumen de sangre que debe expulsarse de las cámaras cardíacas derechas prolonga el cierre de la válvula pulmonar y produce una separación (división fija) de los componentes aórtico y pulmonar del segundo ruido cardíaco. Los niños con defectos auriculares no diagnosticados tienen riesgo de enfermedad vascular pulmonar, aunque esto es raro antes de los 20 años de edad. En casos raros, los lactantes con un cortocircuito grande desarrollan insuficiencia cardíaca congestiva y falta de progreso, lo que obliga al cierre temprano del defecto.
Es improbable que los defectos septales auriculares que miden 8 mm o más se cierren de manera espontánea. Los defectos más pequeños pueden mantenerse en observación en espera del cierre espontáneo en el niño pequeño. Sin embargo, se recomienda el cierre quirúrgico o a través de un catéter en niños con defectos persistentes para reducir el riesgo de largo plazo de enfermedad vascular pulmonar y arritmias auriculares. Tanto el cierre con dispositivo por catéter como el quirúrgico son efectivos y de bajo riesgo. El empleo de la técnica transcatéter depende de la posición y tamaño del defecto. El cierre con esta técnica ha sido muy efectivo para defectos septales de tipo secundario pequeños a medianos y para la persistencia de la ventana oval.
Los defectos en el seno venoso, a menudo relacionados con retorno venoso pulmonar anómalo parcial y defectos del orificio primario, requieren cierre quirúrgico. La cirugía incluye el empleo de circulación extracorpórea e hipotermia leve. La mayoría de los defectos puede cerrarse con el tejido septal natural del paciente o un parche de pericardio o material sintético. La incidencia de secuelas residuales o necesidad de una nueva intervención es muy baja si el cierre se hace durante los primeros 20 años de edad.
Defectos septales ventriculares
Un defecto septal ventricular es una abertura en el tabique interventricular causado por la separación incompleta de los ventrículos durante el desarrollo fetal temprano (figura 32-25B). Estos defectos pueden ser únicos o múltiples y se encuentran en cualquier sitio del tabique interventricular. Los defectos septales ventriculares son la forma más frecuente de cardiopatía congénita, representan del 28% al 42% de todos los casos. La distribución entre varones y mujeres es casi igual. Es posible que el defecto septal ventricular sea la única anomalía cardíaca o que se relacione con múltiples alteraciones cardíacas.
El tabique interventricular tiene 2 orígenes: la hendidura interventricular del corazón tubular plegado que da lugar a la parte muscular del tabique y los cojinetes endocárdicos que se extienden para formar la porción membranosa del tabique. La porción membranosa superior del tabique es la última zona en cerrar, casi siempre para la séptima semana de gestación, y es ahí donde se encuentra la mayoría de los defectos. Los signos y síntomas de estas anomalías dependen del tamaño de la abertura y de la resistencia vascular pulmonar; van desde un soplo asintomático hasta la insuficiencia cardíaca congestiva.
El tamaño del defecto septal ventricular es un factor determinante del cortocircuito de izquierda a derecha, pero no es el único. La magnitud del cortocircuito también depende de la resistencia vascular pulmonar respecto a la resistencia vascular sistémica. En una comunicación pequeña (<5 cm²), la presión más alta del VI impulsa el flujo sanguíneo a la izquierda y el tamaño del defecto limita la magnitud del cortocircuito. La mayoría de los niños con estas anomalías permanece asintomática y tiene bajo riesgo de desarrollar enfermedad vascular pulmonar.
En un cortocircuito más grande y sin restricción (casi siempre >1 cm²), la presión de los ventrículos derecho e izquierdo se iguala y la magnitud del cortocircuito depende de la proporción entre la resistencia vascular pulmonar y la sistémica. En lactantes con grandes defectos septales ventriculares, después del nacimiento la resistencia vascular pulmonar puede mantenerse más alta de lo normal y es factible que en un principio, el tamaño del cortocircuito de izquierda a derecha sea limitado. Conforme continúa el descenso en la resistencia vascular pulmonar en las primeras semanas después del nacimiento por la involución normal de la media de las pequeñas arteriolas pulmonares, crece la magnitud del cortocircuito de izquierda a derecha. Al final se establece un cortocircuito de izquierda a derecha y aparecen las manifestaciones clínicas (ej. taquipnea, diaforesis, sobre todo durante la alimentación, y falta de progreso).
En la mayoría de los casos durante la lactancia, la presión vascular pulmonar sólo se eleva un poco y el principal contribuyente ala hipertensión pulmonar es el aumento en el flujo pulmonar. En algunos lactantes con un defecto septal amplio, el grosor arteriolar pulmonar nunca disminuye. Con la exposición constante al flujo sanguíneo pulmonar elevado, se desarrolla enfermedad pulmonar vascular obstructiva. En pacientes sin tratamiento, la resistencia vascular pulmonar al final rebasa la resistencia sistémica. En este caso, el flujo del cortocircuito se revierte y el niño experimenta cianosis progresiva porque la sangre desoxigenada pasa del lado derecho al lado izquierdo del corazón. Estos síntomas, aunados a los cambios irreversibles en los vasos sanguíneos pulmonares, representan la forma en etapa final de la cardiopatía congénita, llamada complejo de Eisenmenger. Las personas que desarrollan este trastorno tienen una esperanza de vida cercana a 43 años, la causa de muerte es insuficiencia cardíaca progresiva.
El tratamiento de un defecto septal ventricular depende del tamaño de la anomalía, los trastornos hemodinámicos acompañantes y el cuadro clínico. Los niños con defectos pequeños o medianos pueden vigilarse sin intervenir si no aparecen manifestaciones de insuficiencia cardíaca congestiva e hipertensión pulmonar. Los defectos ventriculares no crecen, algunos se cierran de manera espontánea con el tiempo. Por lo general, la ecocardiografía 2D detallada es adecuada para diagnosticar el tamaño y la posición de un defecto, así como para calcular las presiones pulmonares.
Por lo general, el cateterismo cardíaco se reserva para casos en los que es necesario confirmar el grado de reversibilidad de la resistencia vascular pulmonar.
La insuficiencia cardíaca congestiva se trata con medicamentos. Los lactantes sintomáticos a veces requieren suplementos alimentarios o alimentación por sonda para favorecer el crecimiento y el desarrollo. En los lactantes sintomáticos en los que no puede hacerse la reparación completa por el tamaño u otras lesiones que complican el caso, es posible realizar un procedimiento paliativo para disminuir los síntomas. La colocación de una banda sintética alrededor de la arteria pulmonar principal puede reducir el flujo sanguíneo pulmonar hasta que sea factible la reparación completa. El cierre quirúrgico del defecto se completa con la colocación de un parche sintético o autólogo para ocluir la desviación por el tabique ventricular. Estos procedimientos casi siempre se hacen en forma electiva en el lactante o el niño pequeño, sus tasas de morbilidad y mortalidad son bajas. El cierre de los defectos septales ventriculares con un dispositivo a través de un catéter es un área de interés, pero la dificultad para situar con éxito los dispositivos ha limitado su aplicabilidad.
Defectos en el cojinete endocárdico
El conducto AV conecta las aurículas con los ventrículos durante el desarrollo cardíaco temprano. Los cojinetes endocárdicos rodean este conducto y aportan tejido a la parte inferior del tabique interauricular, la parte superior del tabique interventricular, la valva septal de la válvula tricúspide y la valva anterior de la válvula mitral.
Cualquier fallo en el desarrollo de estos tejidos genera un defecto en el cojinete endocárdico. Cerca del 3% de todas las anomalías cardíacas congénitas es un defecto en el cojinete endocárdico, con incidencia casi igual en varones y mujeres. Estos defectos tienen una relación marcada con el síndrome de Down y se encuentran hasta en el 50% de los niños con este síndrome.
Existen diversas variaciones en los defectos del cojinete endocárdico. La anomalía puede describirse como parcial o completa. La anatomía de la válvula AV determina el tipo. En los defectos parciales del conducto AV, los 2 anillos valvulares AV están completos y separados. El tipo más frecuente de anomalías parciales del conducto AV es el del orificio primario, a menudo vinculado con una hendidura en la válvula mitral. En un defecto completo hay un orificio valvular AV común, junto con defectos en el tejido septal auricular y ventricular (figura 32-35E). Otras anomalías cardíacas pueden relacionarse con defectos en el cojinete endocárdico y la mayoría incluye posición anómala cardíaca y tetralogía de Fallot.
Desde el punto de vista fisiológico, los defectos del cojinete endocárdico producen alteraciones similares a las descritas para las anomalías en el tabique auricular o ventricular. La dirección y magnitud de un cortocircuito en un niño con un defecto en el cojinete endocárdico dependen de la combinación de las anomalías y de la resistencia vascular pulmonar y sistémica. Los efectos hemodinámicos de un defecto aislado en el orificio primario son los ya descritos para el defecto septal auricular. Estos niños permanecen casi asintomáticos durante la infancia. Con una anomalía completa en el conducto AV, el flujo sanguíneo pulmonar aumenta cuando la resistencia vascular pulmonar desciende a causa de la desviación de izquierda a derecha a través de los defectos septales ventricular y auricular. Los niños con defectos completos a menudo son intolerantes a los esfuerzos, se fatigan con facilidad, tienen falta de progreso, padecen infecciones recurrentes y otros signos de insuficiencia cardíaca congestiva, sobre todo cuando el cortocircuito es grande. Si la lesión no se corrige, se producen hipertensión pulmonar y aumento en la resistencia vascular pulmonar.
El momento de la corrección de defectos en el cojinete endocárdico depende de la gravedad de la lesión y de los síntomas. En caso de un defecto en el orificio primario, la reparación quirúrgica casi siempre se planifica como un procedimiento electivo antes que el niño llegue a la edad escolar.
La anomalía en el tabique auricular se cierra con un parche y se realiza valvuloplastia mitral, si existe insuficiencia de la válvula. La cirugía correctiva es necesaria para todos los defectos completos del conducto AV. Por lo general, se realiza a edad temprana en el lactante con un parche en ambos defectos septales, ventricular y auricular, además de separación del aparato valvular AV para crear válvulas mitral y tricúspide competentes. Los lactantes con síntomas graves a ameritan un procedimiento paliativo en el que se coloca una banda elástica a la arteria pulmonar principal para reducir el flujo sanguíneo pulmonar. Esto casi siempre mejora la capacidad del lactante para crecer y desarrollarse hasta que pueda realizarse una reparación completa. La reparación quirúrgica total de los defectos completos del conducto AV conllevan un riesgo bajo. En cerca del 11,7% de los niños es necesaria una nueva operación.
Estenosis pulmonar
La obstrucción del flujo sanguíneo del ventrículo derecho a la circulación pulmonar se denomina estenosis pulmonar. La obstrucción puede ocurrir como una lesión valvular aislada, dentro de la cámara ventricular derecha, en las arterias pulmonares o como combinación de estenosis en varias zonas. Es un defecto relativamente frecuente, se calcula que representa cerca del 10% de todos los casos de cardiopatía congénita y a menudo se relaciona con otras anomalías.
Los defectos valvulares pulmonares, el tipo más frecuente de obstrucción, casi siempre producen cierta alteración del flujo sanguíneo pulmonar e incremento del trabajo impuesto al ventrículo derecho (figura 32-25D). La mayoría de los niños con estenosis valvular pulmonar tienen estenosis leve que no se agrava con el tiempo. Estos niños permanecen casi asintomáticos y el diagnóstico se establece por el hallazgo de un soplo sistólico. La estenosis moderada o grave evoluciona con el tiempo, sobre todo antes de los 12 años de edad, por lo que estos niños ameritan un seguimiento cuidadoso. La estenosis pulmonar crítica en el recién nacido se manifiesta con cianosis por el cortocircuito de derecha a izquierda entre las aurículas e hipertensión ventricular derecha.
Estos lactantes requieren prostaglandina E1 para mantener la circulación a los pulmones a través del conducto arterioso.
La valvotomía pulmonar es el tratamiento de elección para todos los defectos valvulares con gradientes de presión mayores de 30 mm Hg entre el ventrículo derecho y la circulación pulmonar.
La valvuloplastia con globo transcatéter ha sido muy exitosa para esta lesión. La estenosis en las arterias pulmonares periféricas también puede tratarse con angioplastia por globo. En fecha reciente se han utilizado endoprótesis vasculares en niños con estenosis arterial pulmonar para mantener los vasos abiertos. Esto se emplea cuando hay fallo en la dilatación con globo.
Tetralogía de Fallot
La tetralogía de Fallot es la cardiopatía congénita cianótica más frecuente.
Como su nombre implica, consiste en 4 defectos relacionados:
- Un defecto septal ventricular que afecta el tabique membranoso y la parte anterior del tabique muscular.
- Dextroposición o desplazamiento a la derecha de la aorta, por lo que queda sobre el ventrículo derecho y se comunica con el defecto septal.
- Obstrucción o estrechamiento de la vía de salida pulmonar, que incluye estenosis valvular pulmonar, reducción de tamaño del tronco pulmonar o ambos.
- Hipertrofia del ventrículo derecho por aumento del trabajo necesario para bombear la sangre por los conductos pulmonares obstruidos (figura 32-25C).
Las variaciones del defecto incluyen cayado aórtico derecho y vena cava superior izquierda persistente. Cuando esto ocurre, el trastorno puede llamarse pentalogía de Fallot.
La cianosis se debe al cortocircuito de derecha a izquierda a través del defecto septal ventricular.
La magnitud de la cianosis depende de la restricción del flujo sanguíneo en el lecho pulmonar. La obstrucción al flujo de salida ventricular derecho hace que la sangre desoxigenada del ventrículo derecho se desvíe por el defecto septal y se expulse a la circulación sistémica. El grado de obstrucción puede ser cambiante, aumenta durante períodos de estrés, lo que causa ataques de cianosis intensa («ataques de tetralogía»). Estas crisis casi siempre ocurren por la mañana durante el llanto, la alimentación o la defecación. Estas actividades aumentan el requerimiento de oxígeno del lactante. El llanto y la defecación pueden además elevar la resistencia vascular pulmonar, lo que intensifica el cortocircuito de derecha a izquierda y reduce el flujo sanguíneo pulmonar. En la crisis cianótica, el lactante experimenta cianosis aguda, hiperpnea, se encuentra irritable y diaforético. Más tarde, el lactante queda lánguido y puede perder la conciencia. La colocación del lactante en una posición genupectoral aumenta la resistencia vascular sistémica, lo que incrementa el flujo sanguíneo pulmonar y reduce el cortocircuito de derecha a izquierda. Durante una crisis de cianosis, los lactantes mayores y los niños a veces asumen la posición en cuclillas de manera espontánea, que funciona como la posición genupectoral para aliviar la crisis. El flujo turbulento por la vía de salida ventricular derecha estrecha produce un soplo sistólico de expulsión áspero característico. La auscultación durante una crisis cianótica revela la disminución o ausencia del soplo debido al descenso drástico en el flujo sanguíneo pulmonar.
Todos los niños con tetralogía de Fallot necesitan la corrección quirúrgica completa. Sin embargo, antes de la operación debe corregirse la anemia ferropénica para prevenir un accidente cerebrovascular. Se mantiene una vigilancia estrecha para detectar deshidratación a fin de prevenir las complicaciones trombóticas, puede administrarse propranolol para prevenir las crisis hipóxicas y, en caso de acidosis, bicarbonato de sodio y agonistas α-adrenérgicos.
Hoy en día se sugiere la reparación definitiva temprana en la lactancia en la mayoría de los centros experimentados en cirugía intracardíaca en lactantes. Cuando existe cianosis extrema en un lactante pequeño o cuando también hay hipoplasia marcada de las arterias pulmonares, a veces es necesario un procedimiento paliativo para facilitar el flujo sanguíneo pulmonar. Esto se hace mediante la colocación de un cortocircuito prostético entre una arteria sistémica y la arteria pulmonar (cortocircuito de BlalockTaussig modificado). La dilatación con globo de la válvula pulmonar también puede resultar paliativa en algunos lactantes. La corrección total se realiza más tarde durante la lactancia o la infancia temprana. La reparación completa incluye cierre con parche del defecto septal ventricular y alivio de cualquier obstrucción a la vía de salida del ventrículo derecho. La reparación se conlleva una tasa de mortalidad menor del 3%, pero los pacientes necesitan seguimiento prolongado para detectar lesiones residuales, dilatación o disfunción ventricular derecha y arritmias. También deben vigilarse porque el riesgo de EI persiste.
Trasposición de grandes arterias
En la trasposición de las grandes arterias, la aorta nace del ventrículo derecho y la arteria pulmonar se origina en el VI (figura 32-25F). La trasposición completa ocurre en uno de cada 4.000 nacidos vivos y es la razón más frecuente de referencia cardiológica pediátrica en las primeras 2 semanas de edad.
La cianosis es el síntoma inicial más frecuente, se produce porque la anomalía permite que la sangre venosa circule por el lado derecho del corazón y se circule de nuevo a los pulmones a través del VI y la arteria pulmonar principal. En lactantes nacidos con este defecto, la supervivencia depende de la comunicación entre los lados derecho e izquierdo del corazón a través del conducto arterioso permeable o un defecto septal. El 50% de los lactantes con trasposición de grandes arterias tiene defectos septales ventriculares, pequeños en el 10%, y permiten la mezcla efectiva de la sangre.
Debe administrarse prostaglandina E1 a los recién nacidos en los que se sospecha esta lesión en un esfuerzo por mantener la permeabilidad del conducto arterioso. Puede recurrirse a la septotomía auricular con globo para aumentar el flujo sanguíneo entre los 2 lados del corazón. En este procedimiento se introduce un catéter con punta de globo en el corazón a través de la vena cava y luego por la ventana oval hasta la aurícula izquierda. A continuación, se infla el globo y se tira de él a través de la ventana oval, con lo que se aumenta el tamaño de la abertura.
La cirugía correctiva es esencial para la supervivencia prolongada. Un procedimiento de cambio arterial, la técnica de elección actual, conlleva tasas de supervivencia mayores del 90%. Es preferible realizar este procedimiento, que corrige la relación de los flujos sanguíneos sistémico y pulmonar, en las primeras 2 o 3 semanas de edad, antes del descenso posnatal en la resistencia vascular pulmonar. Las arterias coronarias se desplazan a la arteria grande izquierda y cualquier defecto septal ventricular se cierra durante el mismo procedimiento. Las complicaciones del procedimiento de cambio arterial incluyen insuficiencia coronaria, estenosis pulmonar supravalvular, insuficiencia neoaórtica y alteraciones de la frecuencia.
Coartación aórtica
La coartación aórtica es un estrechamiento localizado de la aorta, proximal (preductal), distal (posductal) u opuesto a la entrada del conducto arterioso (yuxtaductal, figura 32-25H). Cerca del 98% de las coartaciones son de tipo yuxtaductal. Se cree que la causa de la obstrucción es la constricción de tejido ductal anómalo que se extiende hasta la pared aórtica. La anomalía es más frecuente en varones que en mujeres, con una proporción de hasta 3:1. A menudo se relaciona con otras lesiones cardíacas congénitas, por lo general, válvula aórtica bicúspide en el 46%; además se encuentra en casi el 10% de los sujetos con síndrome de Turner, lo que sugiere un vínculo genético.
El signo típico de la coartación aórtica es la disparidad en las pulsaciones y presión arterial entre brazos y piernas. Las pulsaciones femoral, poplítea y dorsal del pie son débiles o tardías con respecto a los pulsos saltones de los brazos y carótidas. En condiciones normales, la presión arterial sistólica medida en las piernas con el método de manguito es 10 a 20 mm Hg más alta que en los brazos. En la coartación, la presión en las piernas es menor y puede ser difícil de obtener. Los pacientes con coartación a menudo se identifican durante un estudio diagnóstico para hipertensión. La mayoría de las personas con coartación aórtica moderada se mantiene asintomática gracias a los vasos colaterales que se forman alrededor de la zona estrechada. Sin embargo, sin tratamiento la coartación causa hipertensión e hipertrofia del ventrículo izquierdo, con hipertensión sistémica significativa. Los lactantes con coartación grave tienen síntomas tempranos de insuficiencia cardíaca y pueden presentarse en condiciones críticas cuando se cierra el conducto. Si es posible, en estegrupo se requieren la reapertura del conducto arterioso con prostaglandina E1 y cirugía urgente.
Lo ideal es que los niños con coartación que genera un gradiente de presión arterialentre brazos y piernas de 20 mm Hg o más reciban tratamiento antes de los 2 años de edad para disminuir la probabilidad de hipertensión persistente. La corrección quirúrgica suele incluir resección del segmento estrecho de la aorta y anastomosis terminoterminal entre el tejido sano. Por lo general, esto puede hacerse sin circulación extracorpórea, con una tasa de mortalidad cercana a cero. También se ha empleado la angioplastia con globo, con o sin colocación de endoprótesis, aunque la presencia de gradientes residuales y la confiabilidad del acceso quirúrgico han limitado esta técnica. Las complicaciones más frecuentes después de la reparación de la coartación son hipertensión persistente y coartación recidivante. Las tasas de mortalidad quirúrgica aumentan si existe algún defecto relacionado.
Anatomía funcional de ventrículo único
Varias formas de cardiopatía congénita compleja generan un solo ventrículo funcional. Puede haber un ventrículo único derecho o izquierdo, o un ventrículo con morfología indeterminada. La anatomía funcional de ventrículo único es la forma más frecuente de cardiopatía congénita diagnosticada durante la etapa fetal por la incapacidad para obtener la vista de 4 cámaras en la ecografía prenatal de rutina. El síndrome de hipoplasia cardíaca izquierda es la forma más frecuente de anatomía con ventrículo único derecho. La atresia de la válvula tricúspide es la causa más frecuente de un VI único. Se han descrito varias formas más de ventrículo de entrada doble, pero todas las formas de este trastorno tienen efectos patológicos similares y siguen una vía común de intervención.
Todas las formas de anatomía con ventrículo único incluyen una cámara mezcladora común de la sangre venosa pulmonar y sistémica, con grados variables de cianosis. El ventrículo único debe irrigar las circulaciones pulmonar y sistémica. La magnitud del flujo sanguíneo en cada circulación depende de la resistencia en cada sistema. Conforme la resistencia vascular pulmonar cae, se genera preferencia hacia el flujo en la circulación pulmonar y se reduce la circulación sistémica. En algunos defectos, como el síndrome de hipoplasia cardíaca izquierda, el flujo sistémico depende del conducto arterioso permeable. Los recién nacidos con esta lesión casi siempre se presentan con cianosis extrema y síntomas de insuficiencia cardíaca cuando el conducto empieza a cerrarse.
Aunque la anatomía funcional de ventrículo único no puede repararse por completo, la paliación quirúrgica de estos defectos ha sido uno de los logros más innovadores en la intervención para cardiopatía congénita. El objetivo de la paliación quirúrgica es redirigir el retorno venoso directamente a la arteria pulmonar y permitir que el ventrículo único suministre sangre oxigenada a la circulación sistémica. Esto se logra en una serie de 2 o 3 intervenciones quirúrgicas por etapas durante los primeros años de edad del niño. En la actualidad se practica la operación de Fontan y Baudet modificada. El objetivo es evitar el paso por el lado derecho del corazón, por lo que la sangre venosa sistémica se dirige a las arterias pulmonares; esto permite que el ventrículo único bombee a la circulación sistémica. También se recurre al trasplante cardíaco para las formas más complejas de la cardiopatía congénita con ventrículo único (figura 32-26).
Las tasas de supervivencia para los niños con formas complejas de cardiopatía con ventrículo único han mejorado mucho, pero los resultados de largo plazo aún son inciertos. La disfunción ventricular, arritmias y trombosis son muy frecuentes en esta población de pacientes. La definición de las estrategias terapéuticas médicas y quirúrgicas para estos pacientes se mantiene como un área de investigación activa en la cardiología pediátrica y la cirugía cardíaca.
Adultos con cardiopatía congénita
El tratamiento exitoso de la cardiopatía congénita en la población pediátrica ha dado origen a una población creciente de adultos sobrevivientes con diversas lesiones cardíacas congénitas reparadas, no reparadas y paliadas. Un estudio epidemiológico sobre la prevalencia y distribución por edad de la cardiopatía congénita identificó una prevalencia de 6 por cada 1.000 adultos.
Aunque la mayoría de los adultos con cardiopatía congénita se sometió a tratamiento y quizá a cirugía durante la infancia, la mayoría de los defectos cardíacos congénitos debe considerarse como una enfermedad crónica que requiere vigilancia y atención de largo plazo. Sólo las lesiones más sencillas, como la persistencia del conducto arterioso y el defecto septal auricular del tabique secundario no complicado, pueden considerarse reparados por completo. Las preocupaciones fisiológicas crónicas incluyen arritmias, trastornos hemodinámicos, complicaciones de la cianosis prolongada, endocarditis, lesiones residuales y la necesidad de una nueva operación. También es factible que la anomalía cardíaca tenga implicaciones significativas en otros aspectos de la salud, como la tolerancia al ejercicio, cirugía no cardíaca y embarazo. También es preciso considerar varios aspectos psicosociales importantes, como los logros neurocognitivos, empleo, posibilidad de contratar un seguro, planificación familiar, observancia terapéutica y comprensión del trastorno subyacente y los riesgos. La esperanza de vida con algunas de las lesiones más complejas (ej. síndrome de hipoplasia cardíaca izquierda) se desconoce porque los sobrevivientes de mayor edad hasta ahora nacieron en la década de 1980. Ha surgido una nueva especialidad médica creciente para suministrar a los adultos con cardiopatía congénita los servicios especializados que necesitan por parte de médicos que comprendan las complejidades de sus problemas cardíacos y otros aspectos de la atención a la saludo del adulto.
Enfermedad de Kawasaki
La enfermedad de Kawasaki, también conocida como síndrome de ganglio linfático mucocutáneo, es un trastorno febril agudo de niños pequeños. El Dr. Tomisaku Kawasaki fue el primero en describirlo en 1967 en Japón: afecta la piel, cerebro, ojos, articulaciones, hígado, ganglios linfáticos y el corazón. Esta enfermedad es la principal causa de cardiopatía adquirida en niños pequeños, del 15% al 25% de los casos causan aneurismas o ectasias en las arterias coronarias que pueden causar infarto de miocardio, muerte súbita o insuficiencia coronaria crónica. Cada año se hospitalizan más de 4000 niños con enfermedad de Kawasaki en Estados Unidos. Más del 80% de los pacientes con este trastorno tiene 4 años de edad o menos, con una proporción entre varones y mujeres de 1,5:1. Aunque es más frecuente en Japón, la enfermedad afecta niños de muchas etnias, ocurre en todo el mundo y su frecuencia va en aumento.
Patogénesis
La enfermedad se caracteriza por vasculitis (o sea, inflamación de los vasos sanguíneos) que comienza en los vasos pequeños (arteriolas, vénulas y capilares) y avanza hasta afectar algunas de las arterias más grandes, como las coronarias. Se desconoce la causa y patogénesis exactas de la enfermedad, pero se cree que es de origen inmunitario. Durante la fase aguda de la enfermedad se han detectado alteraciones inmunitarias, como aumento en la activación de células T cooperadoras y concentraciones altas de mediadores inmunitarios y anticuerpos que destruyen las células endoteliales. Se formuló la hipótesis de que algún antígeno desconocido, quizá un agente infeccioso frecuente, desencadena la respuesta inmunitaria en un niño con predisposición genética.
Manifestaciones clínicas
La evolución clínica de la enfermedad se describe en 3 fases: aguda, subaguda y convalencencia.
La fase aguda comienza con fiebre de inicio súbito, seguida de conjuntivitis, exantema, compromiso de la mucosa bucal, enrojecimiento e inflamación de manos y pies, además de crecimiento de los ganglios linfáticos cervicales (figura 32-27).
La fiebre casi siempre es alta, alcanza los 40 °C o más, tiene un patrón errático en espigas, no responde a los antibióticos y persiste por 5 días o más. La conjuntivitis es bilateral, comienza poco después de la fiebre, persiste durante todo el período febril y puede durar hasta 4 a 8 semanas. No hay exudado, secreción ni ulceración conjuntival, lo que la distingue de muchos otros tipos de conjuntivitis. Por lo general, el exantema es eritematoso intenso y puede adquirir varias formas, la más frecuente es la urticarial no pruriginosa con grandes placas eritematosas, o un tipo semejante al sarampión. Aunque el exantema casi siempre es generalizado, puede acentuarse en las regiones centrales o periféricas. Algunos niños tienen exantema perianal con distribución en la zona del pañal. Las manifestaciones bucofaríngeas incluyen fisuras en los labios, eritema difuso de la bucofaringe e hipertrofia de las papilas linguales, lo que crea una apariencia en «fresa». Las manos y pies se encuentran inflamados y dolorosos, con enrojecimiento de palmas y plantas. El exantema, las manifestaciones bucofaríngeas y los cambios en manos y pies aparecen 1 a 3 días después del inicio de la fiebre y casi siempre desaparecen cuando ésta cede. El compromiso ganglionar es la característica menos constante de la enfermedad. La adenopatía es cervical y unilateral con un único ganglio linfático crecido y firme, por lo general, mayor de 1,5 cm de diámetro.
La fase subaguda comienza con la defervescencia sintomática y dura hasta que todos los signos de la enfermedad desaparecen. Durante la fase subaguda comienza la descamación de la piel en las puntas de los dedos de manos y pies, y avanza hasta afectar toda la superficie de palmas y plantas. En algunos niños hay descamación en parches en zonas distintas a las manos y pies. La etapa de convalecencia persiste desde la resolución completa de los síntomas hasta que desaparecen todos los signos de inflamación. Por lo general, esto tarda 8 semanas, aunque los cambios inflamatorios en las arterias coronarias pueden persistir hasta por 4 años.
Además de las principales manifestaciones que aparecen en la etapa aguda de la enfermedad, existen varios rasgos menos específicos de la misma que incluyen artritis, uretritis y piuria, manifestaciones gastrointestinales (ej. diarrea, dolor abdominal), hepatitis e hidropesía vesicular.
Existen artritis y artralgia en casi el 30% de los niños con la enfermedad, caracterizadas por inflamación articular simétrica que afecta articulaciones grandes y pequeñas. En casi todos los niños hay compromiso del sistema nervioso central, caracterizado por irritabilidad intensa y labilidad del estado de ánimo.
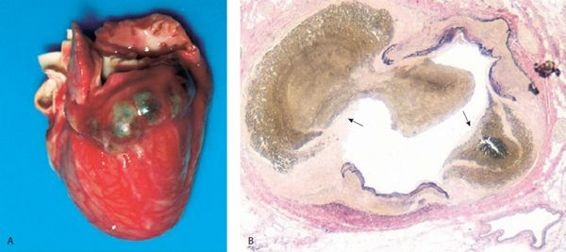
El compromiso cardíaco es la manifestación más importante de la enfermedad de Kawasaki.
Cerca del 15% al 25% de los niños desarrollan alteraciones coronarias, manifestadas por dilatación arterial coronaria y desarrollo de aneurisamas, confirmadas en la ecocardiografía 2D. Las manifestaciones del compromiso arterial coronario incluyen signos y síntomas de isquemia de miocardio y raras veces, infarto de miocardio manifiesto o ruptura de un aneurisma. También puede haber pericarditis, insuficiencia mitral, derrames pericárdicos, miocarditis, endocarditis, insuficiencia aórtica, insuficiencia cardíaca y arritmias. La tasa de mortalidad estimada por enfermedad de Kawasaki es del 2% y, por lo general, se debe al compromiso cardíaco, casi siempre durante la fase de convalecencia de la enfermedad, por trombosis o aneurismas arteriales coronarios.
Diagnóstico y tratamiento
No hay una prueba diagnóstica específica para la enfermedad de Kawasaki, por lo tanto, el diagnóstico se hace con base en el cuadro clínico y acorde con las directrices publicadas. Las directrices sobre persistente al menos por 5 días sin otra causa, acompañada de al menos 4 manifestaciones principales: cambios orales que pueden incluir eritema o fisuras labiales, lengua en fresa y eritema de la mucosa bucal; conjuntivitis bilateral no exudativa; exantema polimorfo, casi siempre troncal,no vesicular; cambios en las extremidades que incluyen eritema y edema de manos y pies, así como descamación de dedos en manos y pies 1 a 3 semanas después del inicio de la enfermedad; y linfadenopatía cervical, a menudo unilateral, con al menos un ganglio con diámetro de 1,5 cm. Las radiografías torácicas, ECG y ecocardiografía 2D se emplean para detectar el compromiso arterial coronario y seguir su evolución. La angiografía coronaria puede emplearse para determinar la magnitud del compromiso arterial coronario.
La gammaglobulina intravenosa (2 g/kg en infusión única) y el ácido acetilsalicílico se consideran las mejores medidas terapéuticas para prevenir las alteraciones coronarias en los niños con enfermedad de Kawasaki. Durante la fase aguda de la enfermedad, el ácido acetilsalicílico casi siempre se administra en dosis altas (80 mg/kg a 100 mg/kg al día dividida en 4 tomas) por sus efectos antiinflamatorios y antipiréticos. Después de controlar la fiebre se reduce la dosis (3 mg/kg a 5 mg/kg al día, dosis única), el fármaco se administra por sus efectos contra la agregación plaquetaria hasta por 8 semanas.
Las recomendaciones para la evaluación cardíaca de seguimiento (pruebas de esfuerzo y a veces angiografía coronaria) se basan en la magnitud de los cambios coronarios. Se recomienda el tratamiento anticoagulante para los niños con aneurismas coronarios múltiples o grandes. Se recomiendan algunas restricciones en las actividades, como los deportes competitivos, en algunos niños con alteraciones coronarias significativas.