03. Miocardiopatías
La definición y clasificación de las miocardiopatías ha evolucionado mucho con el avance de la genética molecular. La definición y clasificación de las miocardiopatías se actualizaron en una declaración científica de la AHA que no sólo incorpora las ventajas de la genética molecular cardíaca, sino otros trastornos recién diagnosticados, además de las conductopatías (canalopatías) iónicas. Esta declaración científica define las miocardiopatías como:
- Un grupo heterogéneo de enfermedades del miocardio relacionadas con disfunción mecánica o eléctrica que casi siempre (aunque no siempre) incluyen hipertrofia o dilatación ventricular inadecuada y que tienen diversas causas, a menudo genéticas.
- Las miocardiopatías se limitan al corazón o forman parte de trastornos sistémicos generalizados, a menudo conducen a la muerte cardiovascular o discapacidad por insuficiencia cardíaca progresiva.
Con base en esta definición, la clasificación de miocardiopatías se divide en 2 grupos principales: primarias y secundarias.
Las miocardiopatías primarias son trastornos cardíacos limitados de miocardio, mientras que las miocardiopatía secundarias son cambios miocárdicos que ocurren en diversos trastornos sistémicos (multiorgánicos). Las miocardiopatías casi siempre se relacionan con trastornos causados por mecanismos mecánicos (ej. insuficiencia cardíaca) o eléctricos (ej. arritmias que ponen en peligro la vida).
Miocardiopatías primarias
Las miocardiopatías primarias se clasifican como genéticas, mixtas o adquiridas, según su etiología.
Las miocardiopatías genéticas incluyen la miocardiopatía hipertrófica (MCH), miocardiopatía ventricular derecha arritmógena (MCDA), miocardiopatía no compactada del ventrículo izquierdo, trastornos hereditarios en el sistema de conducción y conductopatías iónicas.
Las miocardiopatías mixtas, que incluyen MCD, son de origen genético y no genético.
Las miocardiopatías adquiridas incluyen las que tienen un origen inflamatorio (ej. miocarditis), por estrés (pericarditis «tako-tsubo») o por embarazo (miocardiopatía periparto).
En muchos casos se desconoce la causa, en cuyo caso se denomina miocardiopatía idiopática.
Miocardiopatías genéticas
Miocardiopatía hipertrófica
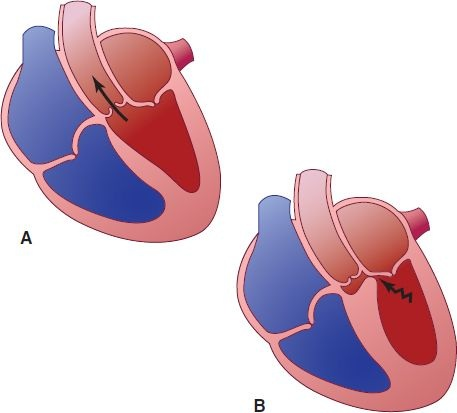
La MCH se caracteriza por hipertrofia ventricular izquierda inexplicable con engrosamiento desproporcionado del tabique interventricular, llenado diastólico anómalo, arritmias cardíacas y en algunos casos, obstrucción intermitente del flujo de salida del ventrículo izquierdo (figura 32-14). Es uno de los tipos más frecuentes de miocardiopatía, ocurre en uno de cada 500 en la población general.
La MCH es la causa más frecuente de MSC en atletas jóvenes. La proclividad a la muerte súbita parece genética y los desfibriladores cardioversores implantables (DCI) han resultados salvadores. Otras complicaciones incluyen fibrilación auricular, accidente cerebrovascular e insuficiencia cardíaca.
La MCH es una enfermedad autosómica dominante causada por mutaciones en los genes que codifican proteínas contráctiles de las sarcómeras cardíacas. En el examen histológico, la MCH incluye hipertrofia de los miocitos con desacomodo de las miofibrillas y aumento de la fibrosis cardíaca. En la actualidad, en las pruebas genéticas pueden identificarse 9 genes vinculados con la MCH, los más frecuentes son los genes de la cadena pesada de la β-miosina y la proteína C de unión con miosina. Se han identificado más de 400 mutaciones individuales y son únicas de una familia a otra. Pueden establecerse algunas relaciones fenotípicas con mutaciones específicas, pero existen muchas excepciones, lo que indica que los modificadores genéticos y los factores ambientales también son importantes. Aunque la MCH es hereditaria, puede evidenciarse en cualquier momento desde la infancia temprana hasta la edad adulta avanzada, con una amplia variedad de manifestaciones y evolución clínica variable.
Las alteraciones fisiológicas básicas de la MCH son disminución del tamaño de la cámara ventricular izquierda, escasa distensibilidad con volumen por latido reducido derivado del llenado diastólico alterado, insuficiencia mitral y en cerca del 25% de los casos, obstrucción dinámica de la salida ventricular izquierda. Las mayoría de los casos puede mantenerse asintomática, pero puede haber disnea, dolor torácico durante el esfuerzo, intolerancia al ejercicio, síncope y arritmias. Debido a la hipertrofia masiva, la presión ventricular izquierda elevada y la alteración potencial de las arterias intramurales, a menudo existe isquemia de miocardio focal, incluso en ausencia de EAC, por lo que el dolor anginoso es frecuente. La MCH a menudo se acompaña de obstrucción del flujo de salida ventricular izquierdo durante el reposo o el esfuerzo, causado por el movimiento anterior sistólico de la válvula mitral y el contacto de la válvula mitral con el tabique ventricular. Las manifestaciones clínicas son muy variables y pueden evolucionar a la insuficiencia cardíaca en etapa terminal con remodelación ventricular izquierda y disfunción sistólica.
El diagnóstico de MCH muchas veces se establece mediante ecocardiografía 2D, que demuestra la hipertrofia ventricular izquierda no dilatada, en ausencia de otras enfermedades cardíacas o sistémicas. La ECG es anómala en el 95% de los casos, muestra hipertrofia ventricular. La vigilancia ambulatoria continua ayuda a detectar arritmias. La IRM cardíaca también ayuda a determinar el sitio y extensión de la hipertrofia. El valor de las pruebas electrofisiológicas es controversial. Las pruebas genéticas, con análisis de secuencia bidireccional del ácido desoxirribonucleico (ADN), proporciona el diagnóstico exacto y permite identificar las mutaciones génicas, si el gen afectado es uno de los 9 que pueden identificarse en las pruebas.
El tratamiento médico de la MCH se enfoca sobre todo en las personas con obstrucción y síntomas. La primera opción para aliviar los síntomas son los medicamentos que bloquean los efectos de las catecolaminas que exacerban la obstrucción al flujo de salida y disminuyen la frecuencia cardíaca para mejorar el llenado diastólico. Por lo general, los bloqueadores β-adrenérgicos son la elección inicial para la MCH sintomática. También puede emplearse el bloqueador del conducto de calcio verapamilo. Sin embargo, puede exacerbar la obstrucción de la salida ventricular izquierda y no se recomienda para personas con obstrucción grave del flujo y síntomas intensos. Puede agregarse disopiramida al β-bloqueador o al verapamilo, ya que tiende a disminuir el gradiente y mejora los síntomas en algunas personas.
En la MCH resistente al tratamiento farmacológico, las alternativas terapéuticas incluyen la miectomía septal, la ablación con alcohol del tabique interventricular y el control de la frecuencia de 2 cámaras y biventricular. La fibrilación auricular se trata con medicamentos que controlan la frecuencia, cardioversión y coagulación. Debe emplearse un DCI en personas con MCH que tienen riesgo de muerte súbita cardíaca. Éstas incluyen a las que ya tuvieron un paro cardíaco, o que experimentaron taquicardia ventricular o fibrilación ventricular, los que tienen un familiar en primer grado con MSC prematura vinculada con MCH, así como aquellos con taquicardia ventricular repetida, grosor >30 mm de la pared ventricular, respuesta hipotensiva al ejercicio y síncope reciente inexplicable. Cerca del 5% de las personas desarrolla MCH en etapa terminal y requieren tratamiento estándar para insuficiencia cardíaca, que incluye la consideración del trasplante cardíaco.
Miocardiopatía/displasia ventricular derecha arritmógena
La miocardiopatía/displasia ventricular derecha arritmógena (C/DVDA) es una enfermedad miocárdica con infiltración fibroadiposa del miocardio ventricular derecho, causa insuficiencia cardíaca derecha y varios trastornos de la frecuencia, en particular taquicardia ventricular. Ocupa el segundo lugar, después de la MCH, como causa de MSC en atletas jóvenes. La incidencia de C/DVDA varía desde uno en 2.000 a uno en 5.000, afecta más a menudo a los varones. Se hereda como rasgo autosómico dominante en el 30% al 50% de los casos, con 8 genes identificados, aunque se han identificado algunas formas recesivas con manifestaciones un poco distintas.
El trastorno se caracteriza por la pérdida progresiva de miocitos, con reemplazo parcial o completo del músculo ventricular derecho por tejido adiposo o fibroadiposo. Este trastorno se relaciona con taquiarritmias ventriculares por reentrada o de origen ventricular derecho, a menudo desencadenadas por la liberación de catecolaminas que causa el ejercicio. Se cree que las manifestaciones clínicas se dividen en 3 fases. En la «fase oculta» temprana, las personas casi siempre permanecen asintomáticas, pero tienen riesgo de MSC, sobre todo durante el esfuerzo. En la «fase eléctrica» el paciente a menudo tiene palpitaciones o síncope. Es durante esta fase que se identifican los cambios ventriculares derechos en la ecocardiografía. En la «enfermedad difusa» siguiente puede haber insuficiencia cardíaca biventricular. Otros síntomas incluyen dolor abdominal y confusión mental.
El diagnóstico de C/DVDA se basa en los hallazgos clínicos, ECG, ecocardiográficos, del monitor Holter, IRM cardíaca, electrocardiografía con señal promediada (ECGSP) e histológicos. Son importantes los antecedentes personales y familiares, incluidos los parientes en primer y segundo grado. Los hallazgos característicos en la ECG de 12 derivaciones incluyen taquicardia ventricular con bloqueo de ramificación izquierda, inversión de la onda T en las derivaciones precordiales derechas y ondas ɛ (pequeñas desviaciones justo después del complejo QRS). También puede haber bloqueo de rama ventricular derecha. Otros estudios diagnósticos que pueden emplearse en la valoración de la C/DVDA incluyen ECG de señal promediada, IRM y angiografía ventricular derecha.
El tratamiento de la C/DVDA se enfoca en la prevención de la MSC. Aunque esta enfermedad no puede curarse, el objetivo terapéutico es controlar la frecuencia cardíaca con antiarrítmicos. A menudo se emplean combinaciones de varios medicamentos. La ablación con radiofrecuencia se emplea en casos resistentes al tratamiento, aunque sólo tiene éxito total en el 30% al 65% de los casos, a veces se requieren múltiples ablaciones. La colocación de un CDI también está indicado en casos resistentes al tratamiento y en los pacientes que sobrevivieron a un episodio de MSC. La colocación de un CDI en otros casos es discutible porque no existe un sistema para estratificación de riesgo. Las opciones terapéuticas finales incluyen ventriculotomía y trasplante cardíaco.
Ventrículo izquierdo no compactado
El ventrículo izquierdo no compactado es una miocardiopatía primaria congénita, al parecer resultado de la embriogénesis anómala, con falta de compactación trabecular del miocardio en desarrollo. Se caracteriza por la distintiva apariencia «esponjosa» del miocardio, sobre todo en la porción apical del VI. Este trastorno puede ser aislado o acompañarse de otras cardiopatías congénitas. Se han identificado casos familiares y no familiares de miocardiopatía no compactada y existen informes de mutaciones en varios genes.
Las manifestaciones derivan sobre todo de las arritmias, fenómenos embólicos e insuficiencia cardíaca. El diagnóstico se hace casi siempre con ecocardiografía 2D y a color, aunque la IRM y la angiografía ventricular izquierda también pueden ser útiles. El tratamiento se enfoca en prevenirlos síntomas de insuficiencia cardíaca, arritmias, accidentes embólicos sistémicos y muerte súbita cardíaca.
Conductopatías iónicas
Los conductos iónicos son proteínas formadoras de poros que constituyen vías para el desplazamiento de iones a través de las membranas celulares. Las enfermedades causadas por mutaciones en genes que codifican las subunidades proteínicas de los conductos iónicos se llaman conductopatías (canalopatías) iónicas. En el corazón, estos trastornos en los conductos iónicos incluyen síndrome de QT largo (SQTL), síndrome de QT corto (SQTC), síndrome de Brugada y taquicardia ventricular polimórfica catecolaminérgica.
EL SQTL y el SQTC se producen por mutaciones en los genes de los conductos para sodio o potasio. El SQTL, que es quizá la conductopatía más frecuente, se identifica en la ECG de 12 derivaciones por el intervalo QT prolongado. Causa una taquicardia ventricular polimórfica conocida como taquicardia helicoidal. El síndrome de QT corto se describió por primera vez en 2000, se caracteriza por intervalo QT corto (<330 ms), lo que puede dar lugar a taquicardia ventricular o fibrilación ventricular y MSC.
El síndrome de Brugada se describió por primera vez en 1992 como una entidad clínica relacionada con una mutación en el gen para el conducto de sodio. Se relaciona con MSC en personas jóvenes, sobre todo varones jóvenes provenientes del sureste asiático que experimentan MSC durante el sueño. En la ECG, el trastorno se caracteriza por bloqueo de rama derecha y elevación del segmento ST en las derivaciones pericárdicas anteriores. La taquicardia ventricular polimórfica catecolaminérgica se produce por un receptor anómalo que regula la liberación de calcio del retículo sarcoplásmico. Se desencadena por la actividad física vigorosa o las emociones intensas y causa síncope, taquicardia ventricular polimórfica y MSC. La ECG de una persona con síndrome de Brugada es característico, incluye un patrón de bloqueo de rama derecha y elevación del segmento ST en V1 a V3.
Miocardiopatías mixtas (genéticas y no genéticas). Miocardiopatía dilatada
La MCD es causa frecuente de insuficiencia cardíaca y la principal indicación para trasplante cardíaco. Se informa que cercan del 20% al 35% es familiar. La mayoría de los casos familiares parecen transmitirse como rasgo autosómico dominante, aunque se han identificado patrones autosómicos recesivos, recesivos ligados a X y mitocondriales. Otras causas incluyen infecciones (virales, bacterianas, micóticas, micobacterianas, parasitarias), toxinas, alcoholismo, fármacos quimioterapéuticos, metales y muchos trastornos más. Con frecuencia no se identifica la causa, en cuyo caso suele llamarse miocardiopatía dilatada idiopática.
La MCD se caracteriza por crecimiento ventricular, disminución del grosor de la pared ventricular y disfunción sistólica de uno o ambos ventrículos (figura 32-15). En el examen histológico se caracteriza por fibras miocárdicas atróficas e hipertróficas, así como fibrosis intersticial. Los miocitos cardíacos, sobre todo los del subendocardio, a menudo muestran cambios degenerativos avanzados. Existe fibrosis intersticial, también más prominente en la región subendocárdica. Puede haber células inflamatorias dispersas.
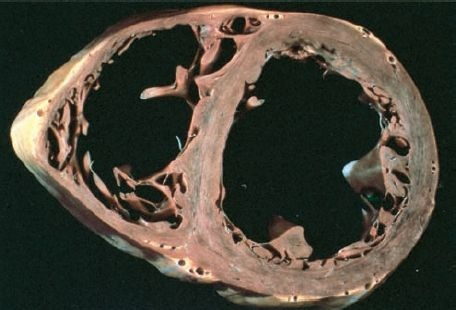
Este trastorno puede manifestarse casi a cualquier edad. Por lo general, se identifica cuando aparecen las manifestaciones clínicas, como disnea, ortopnea y disminución de la capacidad para el ejercicio. En las etapas finales, los pacientes con MCD a menudo tienen fracciones de eyección inferiores al 25% (normal, del 50% al 60%). Conforme la enfermedad avanza, la estasis sanguínea en las paredes de las cámaras cardíacas pueden inducir la formación de trombos y émbolos sistémicos. Son frecuentes la insuficiencia mitral secundaria y la frecuencia cardíaca anómala. Por lo general, la muerte se debe a insuficiencia cardíaca o arritmias, y puede ser súbita.
El tratamiento de la MCD se enfoca en aliviar los síntomas de la insuficiencia cardíaca y reducir el trabajo del corazón. Los fármacos incluyen diuréticos para disminuir la precarga, β-bloqueadores para reducir la frecuencia cardíaca y la demanda miocárdica de oxígeno, fármacos reductores de la poscarga para mejorar la contractilidad y descender las presiones de llenado ventricular izquierdo e inhibidores de la ECA para prevenir la vasoconstricción. También pueden consumirse anticoagulantes para prevenir la formación de trombos y antiarrítmicos. Otros tratamientos incluyen marcapaso biventricular, un DCI biventricular y en casos resistentes al tratamiento, trasplante cardíaco. También es importante eliminar los fármacos causales (si se identifican); evitar los depresores cardíacos, incluido el alcohol; y ajustar el reposo con niveles asintomáticos de ejercicio o actividad.
Miocardiopatía restrictiva primaria
La miocardiopatía restrictiva es una forma rara de enfermedad del músculo cardíaco en la que el llenado ventricular se limita por la rigidez excesiva de las paredes ventriculares. La miocardiopatía restrictiva puede ser idiopática o relacionarse con enfermedades distintivas que afectan el miocardio, sobre todo fibrosis por radiación, amiloidosis, sarcoidosis o tumores metastásicos. También hay informes de factores genéticos que participan en las formas familiares.
Los síntomas de la miocardiopatía restrictiva incluyen disnea, disnea paroxística nocturna, ortopnea, hepatomegalia, edema periférica, ascitis, fatiga y debilidad. Las manifestaciones de esta enfermedad se parecen a las de la pericarditis constrictiva. En la etapa avanzada de la enfermedad existen todos los signos de insuficiencia cardíaca, salvo por la cardiomegalia.
Miocardiopatías adquiridas amatoria
La miocarditis es la inflamación del miocardio, pero su clasificación, diagnóstica y tratamiento son complejos. Las manifestaciones clínicas son muy variables, van desde síntomas inespecíficos como fiebre, mialgias o disnea de esfuerzo, hasta colapso hemodinámico y muerte súbita. La incidencia y prevalencia de la miocarditis es difícil de confirmar por la amplia variación en el cuadro clínico.
Aunque existen varias causas relacionadas con la miocarditis, casi siempre se debe a una infección viral, más a menudo por un enterovirus (virus Coxsackie grupo B). Los adenovirus y parvovirus también están identificados como causa en niños pequeños. Otras etiologías incluyen infecciones bacterianas o micóticas, hipersensibilidad a ciertos fármacos y enfermedades autoinmunitarias, como lupus eritematoso sistémico. La miocarditis es un hallazgo patológico frecuente en pacientes con síndrome de inmunodeficiencia adquirida (sida), aunque no está claro si se debe a la infección misma por el virus de inmunodeficiencia humana o a una infección secundaria.
La miocarditis viral aguda parece evolucionar en 3 fases: la infección viral aguda, activación autoinmunitaria y lesión miocárdica continuada, lo que genera la MCD. Las 3 fases tienen manifestaciones clínicas variables y distintas indicaciones para el tratamiento. Las fases 1 y 2 producen respuestas inflamatorias a la infección viral inicial. Sin embargo, la activación del sistema inmunitario como respuesta a antígenos virales específicos también puede inducir respuestas inflamatorias, independientes de la infección viral, que causan daño a los tejidos del hospedador.
Conforme los leucocitos, linfocitos y macró penetran el miocardio, el edema intersticial y la necrosis focal de miocitos da lugar al reemplazo fibroso. Se ha sugerido que las células T autorreactivas y las citocinas generadas por el hospedador, incluidos el factor de necrosis tumoral α, interleucina-1 e interleucina-6, pueden tener una participación prominente en los cambios miocarditis evolucionan a la fase 3, caracterizada por lesión miocárdica continuada, que al final causa MCD aguda o crónica, insuficiencia ventricular izquierda grave o arritmias que ponen en peligro la vida.
El cuadro clínico de la miocarditis varía desde la ausencia de manifestaciones hasta el shock cardiógeno. Algunos pacientes se presentan con un síndrome viral que incluye fiebre, escalofrío, náuseas, vómito, artralgia y mialgia, que ocurre hasta 6 semanas antes del diagnóstico de miocarditis.
Otras personas se presentan con insuficiencia cardíaca puede ser gradual o súbito y fulminante. Es posible la embolia por el efecto procoagulante de las citocinas combinadas con disminución de la contractilidad miocárdica. En ocasiones el cuadro clínico se parece al SCA, con cambios en el segmento ST y la onda T, marcadores cardíacos positivos y alteraciones en el movimiento regional de la pared, a pesar de que las arterias coronarias son normales. También es factible que los pacientes se presenten con bloqueo AV o cardíaco completo. La miocarditis vital en niños o adultos jóvenes a menudo es inespecífica, con síntomas como fiebre y disminución en el consumo de alimentos.
No existen directrices para la práctica clínica, aunque se han propuesto directrices diagnósticas estandarizadas (criterios Dallas). Estos criterios separan las biopsias iniciales en ausencia de miocarditis, miocarditis limítrofe y miocarditis. Se han identificado varios factores que sugieren que ya no son adecuados.
Los hallazgos de la biopsia endomiocárdica, obtenidos por cateterismo cardíaco, se mantienen como el estándar de referencia para establecer el diagnóstico de miocarditis, a pesar de la exactitud limitada. Otros métodos diagnósticos incluyen el empleo de biomarcadores cardíacos (CK, TnI, TnT) y tinción inmunohistoquímica. Por lo general, la ecocardiografía se realiza en la evaluación inicial por sospecha de miocarditis, aunque los hallazgos pueden ser inespecíficos. Otras técnicas de imágenes cardíacas que están en evaluación incluyen imágenes nucleares con anticuerpos contra miosina marcados con galio o indio, e IRM.
Muchos casos de miocarditis son leves y se autolimitan, por lo que el tratamiento de primera línea es de apoyo. Las medidas iniciales incluyen oxígeno complementario, reposo en cama y antibióticos, en caso necesario. En personas con miocarditis más grave, a veces se requiere apoyo hemodinámico con vasopresores e inotrópicos positivos. Pueden utilizarse inhibidores de la ECA, β-bloqueadores y espironolactona (un antagonista de la aldosterona) para prevenir el deterioro clínico adicional de pacientes con MCD secundaria a miocarditis. Debe considerarse un DCI en sujetos conarritmias documentadas que ponen en peligro su vida. El compromiso de ambos ventrículos casi siempre conlleva un mal pronóstico. El tratamiento inmunosupresor continúa en investigación como alternativa en la miocarditis y no se respalda su utilización habitual.
Miocardiopatía periparto
La miocardiopatía periparto es un raro trastorno del músculo cardíaco que ocurre en el último trimestre del embarazo o los primeros 5 o 6 meses después del parto. Este trastorno es relativamente raro en Estados Unidos, pero en algunas regiones de África se encuentra hasta en el 1% de las embarazadas. La incidencia es mayor en mujeres afroamericanas, multíparas o de edad más avanzada, así como en mujeres con embarazo gemelar, preeclampsia o en las que se emplea tratamiento tocolítico para prevenir el trabajo de parto y parto prematuros.
Aunque se desconoce la etiología de la miocardiopatía periparto, se han propuesto varias causas, incluidas infecciosas inmunitarias, nutricionales, farmacológicas y genéticas. En las biopsias cardíacas de algunas mujeres obtenidas durante la fase sintomática de la enfermedad existen células inflamatorias, lo que sugiere una respuesta inmunitaria alterada. Se manifiesta como la disfunción sistólica VI, con disnea en reposo y de esfuerzo, palpitaciones, edema y ortopnea.
EL diagnóstico de la miocardiopatía periparto puede ser difícil porque los síntomas que pueden ser normales en el embarazo avanzado son similares a las manifestaciones tempranas de la insuficiencia cardíaca. En 1997, en un taller conjunto del National Heart, Lung, and Blood Institute y la Office of Rare Diseases de los National Institutes of Health se identificaron 4 criterios para la definición de la miocardiopatía periparto:
- Insuficiencia cardíaca en el último mes del embarazo o en los 5 meses siguientes al parto.
- Ausencia de una causa identificable de la insuficiencia cardíaca.
- Ausencia de causa identificable de la insuficiencia cardíaca antes del último mes de embarazo.
- Evidencia de disfunción sistólica.
El tratamiento de la miocardiopatía periparto incluye medidas estándar para la insuficiencia cardíaca.
Sin embargo, es necesario considerar los posibles efectos teratógenos y la excreción de medicamentos en la leche materna. El objetivo del tratamiento es reducir la ingestión de líquido y sal; reducir la precarga y la poscarga; aumentar la contractilidad miocárdica; y tratar de prevenir las complicaciones, como la mortalidad. El pronóstico depende de la resolución de la insuficiencia cardíaca. Casi la mitad de las mujeres con miocardiopatía periparto recupera de manera espontánea la función cardíaca normal; la otra mitad queda con disfunción ventricular izquierda que evoluciona a insuficiencia cardíaca manifiesta y muerte temprana.
Miocardiopatía por estrés o «tako-tsubo»
La miocardiopatía por estrés fue descrita por primera vez en Japón, donde ha ocurrido la mayoría de los casos, aunque su incidencia ha aumentado en Estados Unidos. En Japón, se le llamó tako-tsubo, que es una vasija de pesca con cuello estrecho y base amplia que se utiliza para atrapar pulpos. También se ha empleado el término dilatación apical ventricular izquierda transitoria para describir este síndrome.
La miocardiopatía por estrés se identifica en la clínica como disfunción ventricular izquierda transitoria y reversible desarrollada como respuesta al estrés psicológico o emocional intenso. El síndrome se produce sobre todo en mujeres de edad madura que se presentan con IMEST agudo, pero sin evidencia de EAC en el cateterismo cardíaco. Sin embargo, hay compromiso de la contractilidad cardíaca caracterizado por dilatación apical del VI con contractilidad exacerbada en la base del VI.
Se desconoce el mecanismo del aturdimiento miocárdico en la miocardiopatía por estrés,aunque algunas teorías sugieren isquemia por espasmo arterial coronario, espasmo microvascular, predisposición hormonal o lesión directa de los miocitos. Cuando la concentración de catecolaminas se normaliza, el gradiente interventricular se resuelve y la función ventricular izquierda se recupera. El tratamiento es el mismo que para la insuficiencia cardíaca, con un curso corto de anticoagulantes; la mayoría de los pacientes muestra una rápida mejoría y su pronóstico es excelente.
Miocardiopatías secundarias
La miocardiopatía secundaria es una enfermedad del músculo cardíaco en presencia de un trastorno multisistémico (recuadro 32-2). Existen muchos trastornos conocidos que afectan el miocardio; algunos de ellos producen acumulación de sustancias anómalas entre los miocitos (extracelulares), mientras que otras causan acumulación de sustancias anómalas dentro de los miocitos (intracelulares).
Casi 100 enfermedades miocárdicas distintivas pueden causar las manifestaciones clínicas de la MCD. Incluyen miocardiopatías relacionadas con fármacos, diabetes mellitus, distrofia muscular, trastornos autoinmunitarios y fármacos para tratar el cáncer (radiación y fármacos antineoplásicos). La miocardiopatía alcohólica es la causa individual identificable más frecuente de MCD en Estados Unidos y Europa. La doxorrubicina y otras antraciclinas usadas en el tratamiento del cáncer son fármacos potentes cuya utilidad se limita por la toxicidad cardíaca dependiente de la dosis acumulativa. Otro fármacos quimioterapéutico con potencial cardiotóxico es la ciclofosfamida. A diferencia de la lesión primaria en el miocito causada por la doxorrubicina, parece que el principal daño con la ciclofosfamida es vascular, lo que causa hemorragia miocárdica.
Recuadro 32-2. Trastornos relacionados con miocardiopatías secundarias
Trastornos autoinmunitarios:
- Lupus eritematoso sistémico.
- Artritis reumatoide.
- Esclerodermia.
- Poliarteritis nodosa.
Trastornos endocrinos:
- Acromegalia.
- Diabetes mellitus.
- Hipotiroidismo e hipertiroidismo.
- Hiperparatiroidismo.
Enfermedades familiares por almacenamiento
- Enfermedad por almacenamiento de glucógeno.
- Mucopolisacaridosis.
- Hemocromatosis.
Trastornos infiltrativos:
- Amiloidosis.
- Sarcoidosis.
- Fibrosis inducida por radiación.
Trastornos neuromusculares/neurológicos:
- Ataxia de Friedreich.
- Distrofia muscular.
- Neurofibromatosis.
Insuficiencias nutricionales:
- Tiamina (beriberi).
- Proteína (kwashiorkor).
Toxinas:
- Alcohol y sus metabolitos.
- Arsénico.
- Agentes quimioterapéuticos para cáncer (antraciclinas, [doxorrubicina, daunorrubicina], ciclofosfamida).
- Catecolaminas.
- Hidrocarburos.
*No es una lista exhaustiva.