Trastornos de la Función Hepatobiliar y del Pancreas Exocrino
El hígado, la vesícula biliar y el páncreas exocrino se clasifican como órganos accesorios del sistema gastrointestinal. Además de producir secreciones digestivas, el hígado y el páncreas tienen otras funciones importantes. El páncreas endocrino, por ejemplo, aporta la insulina y el glucagón que se requieren para el metabolismo celular, en tanto que el hígado sintetiza glucosa, proteínas plasmáticas y factores de coagulación, y es responsable de la degradación y la eliminación de medicamentos y hormonas, entre otras funciones. Este capítulo se centra en las funciones y los trastornos del hígado, el árbol biliar y la vesícula biliar, así como el páncreas exocrino.
Conceptos clave
Las enfermedades de los hepatocitos alteran las funciones metabólicas y de síntesis del hígado, para generar trastornos en el metabolismo de los carbohidratos, proteínas y grasas; en el metabolismo y la eliminación de fármacos, hormonas, toxinas, amoníaco y bilirrubina de la sangre, y para la interconversión de los aminoácidos y la síntesis de proteínas. El aumento en las concentraciones de aminotransferasas en el suero revela la presencia de daño al hepatocito.
Las enfermedades del sistema de drenaje biliar obstruyen el flujo de la bilis e interfieren con la eliminación de las sales biliares y la bilirrubina para generar daño hepático colestásico, debido a la acumulación de bilis en los lóbulos del hígado. El incremento de la bilirrubina y la fosfatasa alcalina señala la presencia de daño hepático colestásico.
Resumen
Hígado y sistema hepatobiliar
El sistema hepatobiliar está conformado por el hígado, la vesícula biliar y los conductos biliares. El hígado es el órgano más grande del organismo y, en relación con sus funciones, uno de los más versátiles. Se localiza entre el tubo gastrointestinal y la circulación sistémica; la sangre venosa del intestino fluye a través del hígado antes de regresar al corazón. De esta manera, los nutrimentos pueden ser extraídos para su procesamiento y almacenamiento, y las bacterias y otros materiales extraños pueden ser retirados por las células de Kupffer antes de que la sangre regrese a la circulación sistémica.
El hígado sintetiza grasas, glucosa y proteínas plasmáticas.
Otras de sus funciones importantes incluyen la desaminación de los aminoácidos, la conversión del amoníaco en urea, y la interconversión de los aminoácidos y otros compuestos que son fundamentales para los procesos metabólicos del organismo. El hígado produce a diario entre 500 ml y 600 ml de bilis amarilloverdosa. La bilis funge como un vehículo para la excreción de la bilirrubina, el colesterol y ciertos productos del metabolismo orgánico, y contiene sales biliares esenciales para la digestión de las grasas y la absorción de las vitaminas liposolubles. El hígado también retira, conjuga y secreta la bilirrubina hacia la bilis. La ictericia se presenta cuando la bilirrubina se acumula en la sangre. Esto puede presentarse debido a una destrucción excesiva de eritrocitos, incapacidad delhígado para retirar y conjugar la bilirrubina, o a la obstrucción del flujo biliar.
Las pruebas de función hepática, en las que se incluyen las concentraciones séricas de aminotransferasas, se utilizan para valorar la lesión a los hepatocitos.
La bilirrubina sérica, la GGT, la 5’-nucleotidasa y la fosfatasa alcalina se usan como medidas de la función de excreción hepática; la ecografía, la TC y la IRM para evaluar las estructuras hepáticas.
La angiografía puede usarse para visualizar la circulación hepática o portal, mientras que la biopsia hepática permite obtener especímenes tisulares para el análisis microscópico.
Trastornos de la función hepática y biliar
El hígado se encuentra sujeto a la mayoría de los procesos patológicos que afectan a otras estructuras corporales, como los trastornos vasculares, la inflamación, las enfermedades metabólicas, la lesión tóxica y las neoplasias. Como órgano relevante para el metabolismo de fármacos y la destoxificación en el organismo, el hígado está expuesto al daño potencial derivado de una enorme diversidad de fármacos y químicos ambientales. Los medicamentos y los químicos pueden ejercer sus efectos al inducir lesión o muerte en el hepatocito, o por daño colestásico al hígado por lesión de las estructuras para el drenaje biliar. Con base en la estructura química de la sustancia y sus metabolitos, las reacciones medicamentosas pueden ser predecibles o impredecibles (idiosincrásicas) y depender de lascaracterísticas individuales de la persona que la recibe. La identificación temprana de la hepatopatía inducida por fármacos es importante, ya que el retiro de la sustancia resulta curativo en la mayoría de los casos.
La hepatitis se caracteriza por la inflamación del hígado. La hepatitis viral aguda se debe a los virus de la hepatitis A, B, C, D y E.
Si bien todos éstos inducen hepatitis aguda, difieren en cuanto a su forma de transmisión, período de incubación, mecanismo de acción, el grado de daño hepático que producen y su cronicidad, y su capacidad para generar un estado de portador. Las infecciones por VHB, VHC y VHD tienen potencial de evolucionar al estado de portador, a la hepatitis crónica y al carcinoma hepatocelular.
Las enfermedades biliares intrahepáticas alteran el flujo de bilis por el hígado, lo que genera colestasis y cirrosis biliar. Entre las causas de las enfermedades biliares intrahepáticas se encuentran la cirrosis biliar primaria, la colangitis esclerosante primaria y la cirrosis biliar secundaria. Puesto que el alcohol compite por el consumo de cofactores intracelulares que por lo regular se necesitan en el hígado para otros procesos metabólicos, tiende a obstaculizar las funciones metabólicas hepáticas. El espectro de la hepatopatía alcohólica incluye al hígado graso, la hepatitis alcohólica y la cirrosis.
La cirrosis representa la fase terminal de la hepatopatía crónica, en que gran parte del tejido hepático funcional ha sido sustituido por tejido fibroso. El tejido fibroso reemplaza al tejido hepático con funciones normales y produce bandas de constricción que interrumpen el flujo en los canales vasculares y los sistemas de conductos biliares en el hígado.
La disrupción de los canales vasculares predispone a la hipertensión portal y a sus complicaciones, a la pérdida de los hepatocitos y a la insuficiencia hepática eventual. La hipertensión portal se caracteriza por un incremento en la resistencia al flujo de la sangre y al aumento de la presión dentro del sistema venoso porta; las consecuencias patológicas del trastorno incluyen ascitis, formación de canales colaterales para derivación (ej. várices esofágicas) a partir de la circulación portosistémica, y esplenomegalia. La insuficiencia hepática representa la fase terminal de distintas hepatopatías y tiene lugar una vez que menos del 10% al 20% del tejido hepático conserva su funcionalidad.
Las manifestaciones de la insuficiencia hepática corresponden a las distintas funciones del hígado, e incluyen trastornos hematológicos, compromiso de la función endocrina, trastornos cutáneos, síndrome hepatorrenal y encefalopatía hepática.
Existen 2 tipos de cáncer primario en el hígado: el carcinoma hepatocelular (la variante más frecuente, que deriva de los hepatocitos y de sus precursores) y el colangiocarcinoma (cáncer de los conductos biliares, que se origina a partir del epitelio biliar). El carcinoma hepatocelular, que se relaciona con la hepatitis por VHBy VHC, la cirrosis alcohólica y la contaminación de los alimentos (ej. con aflatoxinas) es el quinto tipo de cáncer más común y la tercera causa de mortalidad relacionada con cáncer en el mundo. El colangiocarcinoma se observa sobre todo en adultos mayores con antecedente de trastornos crónicos de los conductos biliares. No obstante, los tumores primarios del hígado son más bien raros en los países desarrollados. El hígado comparte con el pulmón la característica de ser la ubicación más frecuente de los tumores metastásicos.
Trastornos de la vesícula biliar y el páncreas exocrino
Las vías biliares sirven como conducto de paso para la llegada de la bilis desde el hígado hasta el intestino.
Este tracto está conformado por los conductos biliares y la vesícula biliar. Las causas más frecuentes de enfermedad del árbol biliar son la colelitiasis y la colecistitis. Tres factores contribuyen al desarrollo de la colelitiasis: anomalías en la composición de la bilis, estasia de la bilis e inflamación de la vesícula biliar. La cole litiasis predispone la obstrucción del flujo biliar e induce cólico biliar y colecistitis aguda o crónica. El cáncer de la vesícula biliar, que tiene una tasa de sobrevivencia mala a 5 años, se presenta en el 2% de las personas con enfermedad de las vías biliares.
El páncreas es un órgano endocrino y exocrino. El páncreas exocrino sintetiza enzimas digestivas que se secretan en forma inactiva y se transportan hasta el intestino delgado a través del conducto pancreático principal, que suele drenar en el ámpula hepatopancreática y luego en el duodeno, a través del esfínter del conducto pancreático.
Los trastornos más frecuentes del páncreas exocrino son las variantes aguda y crónica de la pancreatitis, así como el cáncer. Las pancreatitis aguda y crónica se relacionan con el reflujo biliar y el alcoholismo crónico. La pancreatitis aguda es una condición inflamatoria del páncreas que se debe a una inactivación inapropiada de las enzimas pancreáticas, con manifestaciones que pueden variar desde las leves hasta las graves y que ponen en riesgo la vida. La pancreatitis crónica causa destrucción progresiva del páncreas endocrino y exocrino. Se caracteriza por episodios de dolor y malestar epigástrico que son similares, pero menos intensos que los que tienen lugar en la pancreatitis aguda. El cáncer del páncreas es la cuarta causa de muerte por cáncer en Estados Unidos. Suele encontrarse muy avanzado en el momento del diagnóstico y su tasa de sobrevivencia a cinco años es del 4%.
Hígado y sistema hepatobiliar
El hígado es el órgano visceral más grande del cuerpo; pesa alrededor de 1,3 kg en el adulto. Se ubica por debajo del diafragma y ocupa gran parte del hipocondrio derecho (figura 46-1).
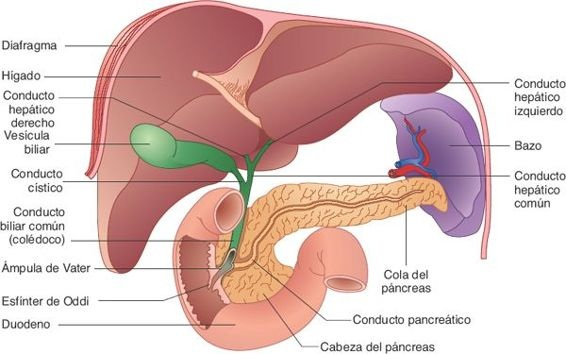
Una cápsula fibroelástica resistente, denominada cápsula de Glisson, lo circunda. Desde la perspectiva anatómica, el hígado se divide en 2 lóbulos grandes (lóbulos derecho e izquierdo) y 2 lóbulos más pequeños (los lóbulos caudado y cuadrado). Excepto por la porción que se ubica en el área epigástrica, el hígado se encuentra contenido por la caja torácica y, por lo regular, no puede palparse en personas saludables.
El hígado recibe el 25% del gasto cardíaco en reposo. Entre los órganos abdominales es el único que cuenta con una irrigación sanguínea doble, constituida por una irrigación venosa (portal) que llega por la vena porta hepática, y una irrigación arterial derivada de la arteria hepática.
Alrededor del 25% de la sangre que ingresa cada minuto al hígado llega por la arteria hepática; el 75% restante ingresa por medio de la vena porta, que carece de válvulas. La sangre venosa que llega por la vena porta hepática proviene del tubo digestivo y de los órganos abdominales principales, entre otros, el páncreas y el bazo (figura 46-2).
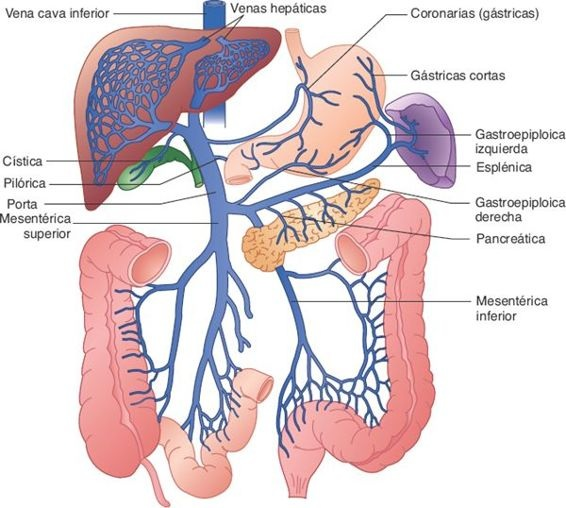
La circulación sanguínea portal lleva nutrimentos y materiales tóxicos que se absorben en el intestino, células hemáticas y sus productos de degradación a partir del bazo e insulina y glucagón del páncreas. Si bien la sangre de la vena porta muestra una saturación incompleta de oxígeno, cubre alrededor del 75% de los requerimientos de oxígeno del hígado.
El flujo venoso de salida del hígado tiene lugar a través de las venas hepáticas que carecen de válvulas y drenan en la vena cava inferior justo por debajo del diafragma. A menudo la diferencia de presión entre la vena hepática y la vena porta es tal, que el hígado tiene capacidad para almacenar alrededor de 500 ml a 1.000 ml de sangre. Esta sangre puede movilizarse hacia la circulación general durante los períodos de hipovolemia y shock. En la insuficiencia cardíaca derecha, cuando la presión en la vena cava aumenta, la sangre retrocede y se acumula en el hígado.
Los lobulillos son las unidades funcionales del hígado. Cada uno es una estructura cilíndrica que mide alrededor de 0,8 mm a 2 mm de diámetro, y varios milímetros de longitud. Existen alrededor de 50.000 a 100.000 lobulillos en el hígado. Cada lobulillo se organiza en torno a una vena central que drena en las venas hepáticas, drenar es correcto y de ahí en la vena cava. Los conductos biliares terminales y las ramificaciones pequeñas de la vena porta y la arteria hepática se ubican en la periferia de lobulillos. Placas de células hepáticas irradian en sentido centrífugo a partir de la vena central, como los radios de una rueda (figura 46-3). Estas placas hepáticas se encuentran separadas por capilares sinusoidales anchos, de pared delgada, denominado sinusoides, que se extienden de la periferia de lobulillos hacia su vena central.
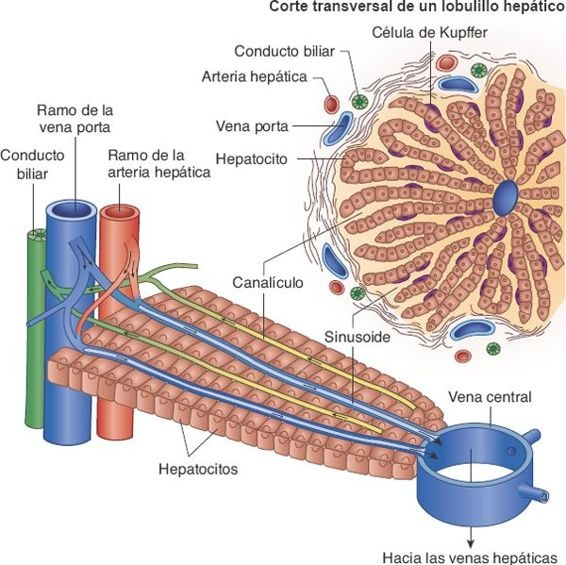
Los sinusoides reciben irrigación de la sangre de la vena porta y de la arteria hepática. Se encuentran en contacto íntimo con las células hepáticas, y participan en el intercambio de sustancias entre la sangre y los hepatocitos. Están recubiertos con 2 tipos de células: las células endoteliales capilares típicas y las células de Kupffer. Estas últimas son células reticuloendoteliales capaces de eliminar y fagocitar células hemáticas viejas y defectuosas, bacterias y otros materiales extraños que llegan en la sangre portal al tiempo que fluyen por el sinusoide. Esta acción fagocítica elimina los bacilos entéricos y otras sustancias lesivas que se filtran hacia la sangre a partir del intestino.
Una importante función exocrina del hígado es la secreción de bilis. Pequeños canales tubulares, denominados canalículos biliares, ubicados entre las membranas celulares de los hepatocitos adyacentes, también suplen a los lóbulos. La bilis que producen los hepatocitos fluye hacia los canalículos y luego hacia la periferia de los lobulillos, para drenar en conductos cada vez mayores, hasta que llegan a los conductos hepáticos derecho e izquierdo. Con frecuencia, los conductos biliares intrahepáticos y extrahepáticos se denominan, en conjunto, árbol hepatobiliar. Estos conductos se unen para constituir el conducto biliar común, que tiene alrededor de 10 cm a 15 cm de longitud, se orienta en dirección caudal, pasa por detrás del páncreas e ingresa al duodeno. El conducto pancreático se une al conducto biliar común en un conducto corto dilatado llamado ámpula hepatopancreática (ámpula de Vater), que drena en el duodeno a través de la papila duodenal. El tejido muscular en la unión de la papila, que en ocasiones se denomina esfínter de Oddi, regula el flujo de bilis hacia el duodeno. Cuando este esfínter se encuentra cerrado, la bilis se desplaza en dirección retrógrada hacia el conducto biliar común y la vesícula biliar.
Funciones metabólicas del hígado
El hígado es uno de los órganos más versátiles y activos del organismo. Produce la bilis, metaboliza hormonas y fármacos; sintetiza proteínas, glucosa y factores de la coagulación; almacena vitaminas y minerales; transforma en urea el amoníaco que se produce por la desaminación de los aminoácidos,y convierte los ácidos grasos en cetonas. También degrada los nutrimentos excedentes y los convierte en sustancias esenciales para el organismo. En su capacidad para metabolizar los fármacos y las hormonas, el hígado funge como un órgano excretor. En este sentido, la bilis, que transporta los productos finales de las sustancias que se metabolizan en el hígado, es en gran medida como la orina, que lleva los desechos corporales que filtran los riñones. Las funciones del hígado se resumen en la tabla 46-1.
Metabolismo de carbohidratos
El hígado desempeña un papel esencial en el metabolismo de los carbohidratos y la homeostasis de la glucosa (figura 46-4).
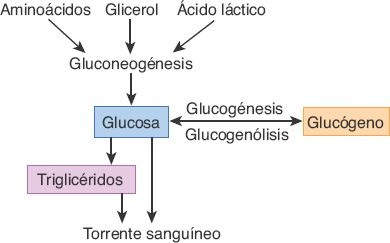
Los hepatocitos tienen la capacidad de almacenar grandes cantidades de glucosa en forma de glucógeno, mediante un proceso denominado glucogénesis. Cuando las concentraciones de glucosa en la sangre son bajas, el glucógeno vuelve a convertirse en glucosa por medio de la glucogenólisis, proceso en que participa una enzima fosfatasa específica de las células hepáticas. El hígado también produce glucosa a partir de los aminoácidos, el glicerol y el ácido láctico, como medio para mantener la glucemia durante los períodos de ayuno o incremento de la demanda. El hígado, además, convierte el exceso de carbohidratos en triglicéridos, para permitir su almacenamiento en el tejido adiposo.
Síntesis de proteínas y conversión del amoníaco en urea
El hígado es un sitio importante para la síntesis y degradación de las proteínas. Produce las proteínas para cubrir sus propios requerimientos celulares, y también las proteínas secretoras que se liberan hacia la circulación. La albúmina es la más importante de las proteínas secretoras; contribuye de manera significativa a la presión coloidosmótica del plasma, así como a la unión y el trasporte de numerosas sustancias, entre éstas, algunas hormonas, ácidos grasos, bilirrubina y otros aniones. El hígado produce asimismo otras proteínas importantes, como el fibrinógeno y los factores de la coagulación.
Mediante distintos procesos anabólicos y catabólicos, el hígado es el sitio principal para la interconversión de aminoácidos (figura 46-5).
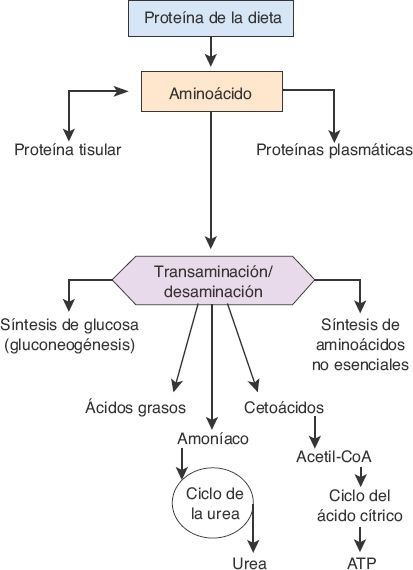
El catabolismo y la degradación en el hígado implican 2 reacciones básicas: la transaminación y la desaminación. En la transaminación se transfiere un grupo amino (NH2) a una sustancia aceptora. Como consecuencia de la transaminación, los aminoácidos pueden participar en el metabolismo intermedio de los carbohidratos y los lípidos.
Durante los períodos de ayuno o inanición, los aminoácidos se utilizan en la producción de glucosa (es decir, la gluconeogénesis). La mayor parte de los aminoácidos no esenciales se sintetiza en el hígado mediante transaminación, un proceso que es catalizado por las aminotransferasas, enzimas que se encuentran en grandes cantidades en este órgano.
La desaminación oxidativa implica el retiro de grupos amino a partir de los aminoácidos, y la conversión de estos últimos en cetoácidos y amoníaco. Esto sucede sobre todo mediante transaminación, en la que se retiran grupos amino y luego se transfieren a otra sustancia aceptora que, entonces, puede trasladar el grupo amino a otra sustancia más, o liberarlo en forma de amoníaco. Dado que el amoníaco es muy tóxico para los tejidos corporales, en particular para las neuronas, el que se libera durante la desaminación se retira de la sangre en el hígado con rapidez y se convierte en urea. En esencia, toda la urea que se forma en el organismo se obtiene por medio del ciclo de la urea en el hígado, y luego se excreta, sobre todo, a través de los riñones. Cierta cantidad se difunde hacia el intestino, donde se convierte en amoníaco por la acción de las bacterias entéricas.
La producción intestinal de amoníaco también deriva de la desaminación bacteriana de los aminoácidos y las proteínas de la dieta que no se absorben, de las células exfoliadas o de la sangre en el tubo gastrointestinal. Este amoníaco pasa a la circulación portal y se transporta hacia el hígado, donde se convierte en urea antes de alcanzar la circulación sistémica. La producción intestinal de amoníaco se incrementa después de la ingestión de alimentos ricos en proteínas y con la hemorragia gastrointestinal. En la hepatopatía avanzada es frecuente el compromiso de la síntesis de urea, lo que origina la acumulación del amoníaco en la sangre.
Vías para el metabolismo lipídico
Si bien la mayoría de las células del organismo metaboliza los lípidos, algunos aspectos de este metabolismo tienen lugar sobre todo en el hígado, lo que incluye la oxidación de los ácidos grasos libres para obtener cetoácidos que aportan energía para otras funciones corporales; la síntesis del colesterol, los fosfolípidos y las lipoproteínas, y la formación de triglicéridos a partir de carbohidratos y proteínas (figura 46-6).
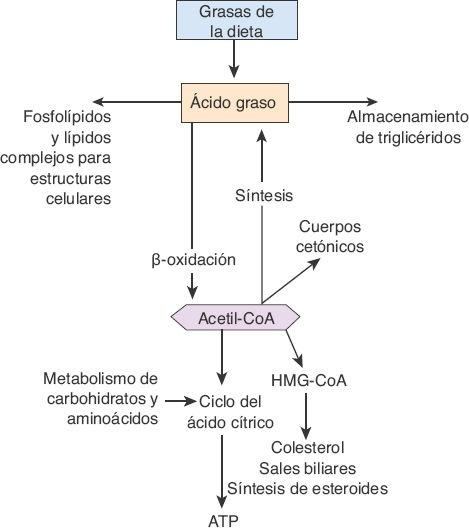
Para obtener energía a partir de los triglicéridos, la molécula debe, primero, dividirse en glicerol y ácidos grasos, para luego escindir a los ácidos grasos en unidades de 2 carbonos de acetilcoenzima A (acetil-CoA) mediante un proceso denominado β-oxidación. El acetil-CoA se canaliza con facilidad hacia el ciclo del ácido cítrico para generar trifosfato de adenosina (ATP). Dado que el hígado no puede utilizar todo el acetil-CoA que se forma, convierte el exceso en ácido acetoacético, un cetoácido muy soluble que se libera hacia el torrente sanguíneo y se transporta hacia otros tejidos, donde se usa para obtener energía. Durante los períodos de inanición, las cetonas se convierten en una fuente importante de energía, al tiempo que los ácidos grasos liberados a partir del tejido adiposo se convierten en cetonas en el hígado.
Las unidades de acetil-CoA derivadas del metabolismo de las grasas también se emplean para sintetizar colesterol y ácidos biliares en el hígado. En este órgano, el colesterol puede utilizarse en distintas formas: puede esterificarse y almacenarse; puede exportarse unido a lipoproteínas, o puede convertirse en ácidos biliares. El paso limitante de la velocidad para la síntesis del colesterol es el que cataliza la reductasa de la 3-hidroxi-3-metil-glutaril-coenzima A (reductasa de la HMG-CoA).
Los inhibidores de la reductasa de la HMG-CoA, o estatinas (fluvastatina, lovastatina, pravastatina y atorvastatina), se utilizan para controlar las concentraciones elevadas de colesterol, al inhibir este paso en la síntesis de la sustancia.
En el organismo, casi toda la síntesis de lípidos a partir de carbohidratos y proteínas tiene lugar en el hígado. Así, siempre que ingresa una cantidad de carbohidratos mayor a la que puede utilizarse de inmediato, el exceso se convierte en triglicéridos en el hígado. Estos triglicéridos se transportan sobre todo en lipoproteínas de baja densidad (LBD) hacia el tejido adiposo, donde se almacenan.
Producción de bilis y colestasis
La secreción de bilis es esencial para la digestión de las grasas de la dieta y la absorción de éstas y de las vitaminas liposolubles a partir del intestino. El hígado produce a diario alrededor de 500 ml a 600 ml de bilis de color amarillo-verdoso. La bilis contiene agua, sales biliares, bilirrubina, colesterol y ciertos productos colaterales del metabolismo. De éstos, sólo las sales biliares, que se forman a partir del colesterol, son importantes para la digestión. Los otros componentes dependen de la secreción de sodio, cloruro, bicarbonato y potasio en los conductos biliares.
Las sales biliares tienen un papel importante en la digestión; facilitan la emulsificación de las grasas de la dieta, y son necesarias para la formación de los micelios que transportan a los ácidos grasos y a las vitaminas liposolubles hacia la superficie de la mucosa intestinal para su absorción. El sistema para la recirculación de la bilis, la circulación enterohepática, incluye componentes múltiples. El hígado, el árbol biliar, la vesícula biliar, la circulación venosa portal, el intestino delgado, el colon y los riñones, desempeñan algún papel en grado variable. Más del 90% de las sales biliares que ingresan al intestino se reabsorbe hacia la circulación portal por medio de un proceso de transporte activo que tiene lugar en la región distal del íleon. A partir de la circulación portal, las sales biliares se desplazan hacia el interior de las células hepáticas, y se reciclan. Por lo general, las sales biliares pasan por todo este circuito alrededor de 17 veces antes de ser expulsadas en las heces.
Colestasis
La colestasis representa una disminución del flujo biliar por los canalículos intrahepáticos, y una reducción en la secreción de agua, bilirrubina y ácidos biliares a partir de los hepatocitos. Como consecuencia, los materiales que por lo regular se transfieren hacia la bilis, entre otros la bilirrubina, el colesterol y los ácidos biliares, se acumulan en la sangre. La condición puede derivar de una hepatopatía intrínseca, en cuyo caso se denomina colestasis intrahepática, o de la obstrucción de los conductos biliares mayores, que origina lo que se conoce como colestasis extrahepática.
Distintos mecanismos están implicados en la patogénesis de la colestasis. La cirrosis biliar primaria (un trastorno autoinmunitario) y la colangitis esclerosante primaria se deben a trastornos de los canalículos intrahepáticos pequeños y los conductos biliares. En el caso de la obstrucción extrahepática, que puede derivar de afecciones como la colelitiasis, las estenosis del conducto biliar común o de las neoplasias que generan obstrucción, los efectos comienzan con un incremento de la presión dentro de los conductos biliares grandes. Los trastornos genéticos que pueden producir colestasis incluyen la colestasis benigna recurrente, el síndrome de Byler y el síndrome de Alagille.
La colestasis benigna recurrente afecta al transporte de bilis hacia los canalículos. El síndrome de Byler también se conoce como colestasis intrahepática familiar progresiva tipo I. La mutación genética responsable de la enfermedad induce diarrea, prurito e insuficiencia hepática. El gen también se ubica en el intestino delgado y el páncreas, y afecta los sistemas gastrointestinal y endocrino. El síndrome de Alagille es un trastorno autosómico dominante que da origen a una hipoplasia intrahepática que afecta de manera específica los conductos biliares interlobares. Los pacientes con el síndrome presentan anomalías cardíacas y oftálmicas aunadas a malformaciones esqueléticas, de manera específica en los huesos faciales.
Las características morfológicas de la colestasis varían según la causa subyacente. La acumulación de pigmento biliar en el hígado es común a todos los tipos de colestasis obstructiva y hepatocelular. En los canalículos biliares dilatados pueden observarse tapones elongados de bilis de color verde-pardo. La ruptura de los canalículos conduce a la extravasación de la bilis y a cambios degenerativos subsecuentes en los hepatocitos circundantes. La colestasis obstructiva prolongada no sólo determina cambios grasos en los hepatocitos, sino la destrucción del tejido conectivo de soporte, lo que da origen a reservorios de bilis que contienen de tritos celulares y pigmento. La obstrucción no resuelta conduce a la fibrosis de las vías biliares y, por último, a la cirrosis biliar en fase terminal.
El prurito es el síntoma más frecuente en las personas con colestasis, y quizá esté relacionado con un incremento de los ácidos biliares en el plasma. Pueden desarrollarse xantomas cutáneos (acumulaciones localizadas de colesterol), resultado de la hiperlipidemia y de las anomalías para la excreción del colesterol. Un hallazgo de laboratorio característico es la elevación en la concentración sérica de fosfatasa alcalina, una enzima presente en el epitelio del conducto biliar y la membrana canalicular de los hepatocitos. Otras manifestaciones de la disminución del flujo biliar se asocian con la absorción intestinal, e incluyen insuficiencias nutricionales de las vitaminas liposolubles A, D y K.
Eliminación de bilirrubina e ictericia
La bilirrubina es el producto final de la degradación del grupo hem que contienen los eritrocitos viejos. Es la sustancia que confiere a la bilis su color. En el proceso de degradación, la hemoglobina que deriva del eritrocito se degrada para formar biliverdina, que se convierte con rapidez en bilirrubina libre (figura 46-7). Ésta, que es insoluble en el plasma, se transporta en la sangre unida a la albúmina plasmática, pero incluso cuando se da esta unión se sigue nombrando bilirrubina libre, para distinguirla de la bilirrubina conjugada. Al tiempo que pasa por el hígado, la bilirrubina libre se traslada a través de la membrana de los hepatocitos y se libera de su molécula de albúmina portadora.
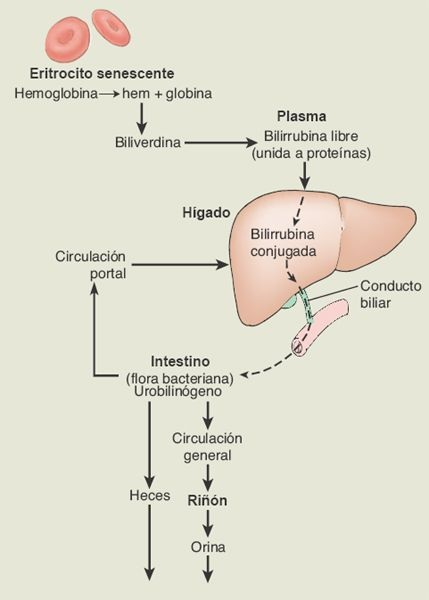
Dentro de los hepatocitos, la bilirrubina libre se convierte en bilirrubina conjugada, lo que la hace soluble en la bilis. La bilirrubina conjugada se excreta como un constituyente de la bilis, y en esta forma pasa por los conductos biliares hacia el intestino delgado donde, debido a la acción de la flora intestinal, cerca de la mitad de la bilirrubina se convierte en una sustancia muy soluble denominada urobilinógeno. Alrededor de una quinta parte del urobilinógeno que se produce se absorbe hacia la circulación portal, y el remanente se excreta en las heces. La mayor parte del urobilinógeno que se absorbe regresa al hígado para volver a excretarse hacia la bilis.
Por lo general, en la sangre sólo se detecta una cantidad escasa de bilirrubina; la concentración normal de bilirrubina sérica total es menor de 1,5 mg/dl (17 μmol a 20,5 μmol). Las mediciones de bilirrubina realizadas en el laboratorio suelen cuantificar tanto la bilirrubina libre como la conjugada, y también la bilirrubina total. Estos resultados se informan como bilirrubina directa (conjugada) y bilirrubina indirecta (no conjugada o libre).
Ictericia
La ictericia, o la pigmentación amarillenta de la piel y los tejidos profundos, deriva de las concentraciones elevadas anómalas de bilirrubina en la sangre. Se desarrolla cuando existe un desequilibrio entre la síntesis de bilirrubina y su eliminación. La ictericia se hace evidente cuando las concentraciones de bilirrubina sérica se elevan por encima de 2 mg/dl a 2,5 mg/dl (34,2 μmol a 42,8 μmol) 5,10 . Debido a que la piel normal tiene un tono amarillo, los signos tempranos de la ictericia son muchas veces difíciles de detectar, en particular en personas de piel oscura. La bilirrubina tiene afinidad singular por el tejido elástico. La esclerótica del ojo, que contiene una proporción alta de fibras elásticas, suele ser una de las primeras estructuras en que puede detectarse la ictericia (figura 46-8).
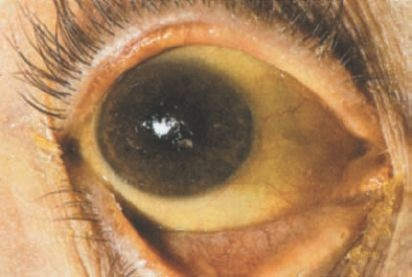
Las 5 causas principales de ictericia son la destrucción excesiva de eritrocitos, las anomalías en la captación de la bilirrubina en los hepatocitos, la disminución de la conjugación de la bilirrubina, la obstrucción al flujo biliar en los canalículos de los lobulillos hepáticos o en los conductos biliares intrahepáticos o extrahepáticos, y la excesiva síntesis extrahepática de bilirrubina. Desde el punto de vista anatómico, la ictericia puede clasificarse como prehepática, intrahepática y poshepática. El cuadro 46-1 enumera las causas comunes de estos tipos de ictericia.
La causa primordial de ictericia prehepática es la hemólisis excesiva de los eritrocitos. La ictericia hemolítica se desarrolla cuando los eritrocitos se destruyen a una velocidad que excede la capacidad del hígado para eliminar la bilirrubina de la sangre. Puede presentarse tras una reacción hemolítica posterior a una transfusión sanguínea, por una disminución del período de vida de los eritrocitos donados, o en trastornos como la esferocitosis hereditaria, en la que las membranas eritrocitarias son defectuosas, o bien en la enfermedad hemolítica del recién nacido. Cuando se presenta una hemorragia interna también puede existir una producción excesiva de bilirrubina al tiempo que tiene lugar la reabsorción de la sangre. Además, los trastornos que generan una eritropoyesis ineficaz pueden incrementar la generación de bilirrubina. La hiperbilirrubinemia neonatal se debe a un aumento en la producción de bilirrubina en los recién nacidos y a su capacidad limitada para excretarla entre los días 0 y 14 de vida. Los neonatos prematuros se encuentran en un riesgo particu lar debido a que sus eritrocitos tienen una vida más corta y una tasa de recambio mayor. En la ictericia prehepática el trastorno pigmentario es leve, se eleva la bilirrubina no conjugada, las heces tienen coloración normal y no se detecta bilirrubina en la orina.
La ictericia intrahepática o hepatocelular se debe a trastornos que afectan de manera directa la capacidad del hígado para retirar la bilirrubina de la sangre o conjugarla, de tal forma que pueda ser eliminada en la bilis. La enfermedad de Gilbert se hereda como rasgo dominante y provoca una disminución, del 66% en promedio, del retiro de la bilirrubina de la sangre. El trastorno es benigno y bastante común, con una tasa de prevalencia aproximada del 8%. Las personas afectadas son asintomáticas, excepto por una concentración un poco elevada de bilirrubina no conjugada e ictericia leve. La conjugación de la bilirrubina se encuentra comprometida siempre que las células hepáticas se dañan, cuando el trasporte de bilirrubina hacia el interior de los hepatocitos se vuelve deficiente o cuando se carece de las enzimas necesarias para conjugar la bilis. Las hepatopatías, como la hepatitis y la cirrosis, son las causas más frecuentes de ictericia intrahepática. Los fármacos, como el anestésico halotano, anticonceptivos orales, estrógenos, esteroides anabólicos, isoniazida,rifampicina y clorpromazina también pueden estar implicados en este tipo de ictericia. La ictericia intrahepática o hepatocelular suele interferir con todas las fases del metabolismo de la bilirrubina: captación, conjugación y excreción. Tanto la bilirrubina conjugada como la no conjugada se encuentran elevadas; con frecuencia la orina es oscura debido a la presencia de bilirrubina en ella, y la fosfatasa alcalina sérica muestra un aumento ligero.
La ictericia poshepática u obstructiva, también denominada ictericia colestásica, tiene lugar cuando existe obstrucción al flujo biliar entre el hígado y el intestino; la obstrucción podría ubicarse en cualquier punto entre la unión del conducto hepático derecho o izquierdo y el sitio en que el conducto biliar se abre en el intestino. Entre las causas se encuentran la estenosis del conducto biliar, los litos vesiculares y los tumores del conducto biliar o del páncreas. La bilirrubina conjugada suele mostrar elevación; las heces tienen color blanquecino debido a la carencia de bilirrubina en la bilis; la orina es oscura; las concentraciones séricas de fosfatasa alcalina muestran incremento marcado, y los niveles de aminotransferasa muestran aumento discreto. Las concentraciones sanguíneas de ácidos biliares suelen ser altas en la ictericia obstructiva. Al tiempo que los ácidos biliares se acumulan en la sangre, se desarrolla prurito. El prurito que precede a la ictericia es común en la ictericia obstructiva.
Pruebas de función hepatobiliar
En la mayoría de los casos, la anamnesis y exploración física aportan información clave sobre la función hepática. Las pruebas diagnósticas ayudan a evaluar la actividad del hígado y el grado de daño que presenta el órgano. Las pruebas de laboratorio se utilizan con frecuencia para valorar la función hepática y confirmar el diagnóstico de hepatopatía.
Las pruebas de función hepática, que incluyen las concentraciones séricas de enzimas hepáticas, se usan para facilitar el diagnóstico de la enfermedad, diferenciar entre los distintos trastornos, determinar la gravedad del padecimiento existente y vigilar las respuestas al tratamiento establecido. Los valores elevados en las pruebas de enzimas séricas suelen indicar la existencia de lesión hepática antes que otros indicadores de la función hepática. Las enzimas clave son la aminotransferasa de alanina (ATL) y la aminotransferasa de aspartato (ATS), que se encuentran en todos los hepatocitos. La ATL es específica del hígado, en tanto que la ATS deriva de órganos distintos al hígado. En la mayor parte de los casos de daño hepático existen incrementos paralelos de la ATL y la ATS. El aumento más radical se observa en los casos de lesión hepatocelular aguda, como sucede en la hepatitis viral, la lesión hipóxica o isquémica, la lesión tóxica aguda o el síndrome de Reye.
La capacidad de síntesis del hígado se refleja en las mediciones de las concentraciones de proteínas séricas y el tiempo de protrombina (es decir, síntesis de factores de la coagulación). La hipoalbuminemia secundaria a la depresión de la síntesis puede complicar la hepatopatía grave. Suelen presentarse insuficiencias de los factores de la coagulación V y los dependientes de la vitamina K (II, VII, IX y X).
La bilirrubina sérica, la gammaglutamiltransferasa (GGT), la 5’-nucleotidasa y la fosfatasa alcalina miden la función excretora del hígado. La fosfatasa alcalina y la 5’-nucleotidasa se identifican en las membranas ubicadas entre los hepatocitos y el conducto biliar, y son liberadas por trastornos que afectan al conducto biliar. La GGT se localiza en el retículo endoplásmico de los hepatocitos y en las células epiteliales del conducto biliar. Se cree que participa en el transporte de los aminoácidos y los péptidos hacia el interior de los hepatocitos. La cuantificación de la GGT puede resultar útil para el diagnóstico de consumo excesivo de alcohol y es un indicador de enfermedad hepatobiliar.
La ecografía aporta información sobre el tamaño, la composición y el flujo sanguíneo del hígado. Ha sustituido en gran medida a la colangiografía para la detección de litos en la vesícula biliar o el árbol biliar. La tomografía computarizada (TC) aporta información similar a la que se obtiene con la ecografía. Las imágenes por resonancia magnética (IRM) han probado ser útiles en algunos trastornos. La angiografía selectiva de las arterias celíaca, mesentérica superior o hepática, puede utilizarse para visualizar la circulación hepática o portal. Una biopsia hepática constituye un medio para analizar el tejido hepático sin cirugía. Existen varias técnicas para obtener tejido hepático: biopsia hepática percutánea, que recurre a una aguja para succión, corte, o corte de carga con resorte; biopsia hepática laparoscópica, y biopsia con aguja fina, que se lleva a cabo con guía ultrasonográfica o con TC. El método a utilizar depende del número de especímenes que se requiere y de la cantidad de tejido necesario para la evaluación. La biopsia hepática laparoscópica constituye un medio para examinar las masas abdominales, evaluar la ascitis de causa desconocida y realizar el estadiaje de los cánceres hepáticos.
Trastornos de la función hepática y biliar
Las estructuras del sistema hepatobiliar se encuentran sujetas a muchas de las mismas condiciones patológicas que afectan a otros sistemas corporales: lesión por medicamentos y toxinas; infección, inflamación y respuestas inmunitarias; trastornos metabólicos y neoplasias. Esta sección se centra en las alteraciones de la función hepática que derivan de la lesión inducida por fármacos; la hepatitis viral y autoinmunitaria; los trastornos intrahepáticos del árbol biliar; la enfermedad hepática inducida por alcohol; la cirrosis, la hipertensión portal y la insuficiencia hepática; y el cáncer hepático.
Trastornos por hepatotoxicidad
En virtud de sus muchos sistemas enzimáticos implicados en las transformaciones y las modificaciones bioquímicas, el hígado desempeña un papel importante en el metabolismo de muchos fármacos y sustancias químicas. El hígado es en particular importante en función del metabolismo de sustancias liposolubles que no pueden excretarse de manera directa a través de los riñones. Es central para la disposición metabólica de casi todos los medicamentos y las sustancias extrañas. Por ende, la toxicidad hepática inducida por fármacos es una complicación potencial del consumo de muchos medicamentos.
Metabolismo de fármacos y hormonas
Se reconocen 3 tipos principales de reacciones que están implicadas en la destoxificación hepática y el metabolismo de los fármacos y otros químicos:
- Reacciones de fase 1, que implican la modificación o la inactivación química de una sustancia.
- Reacciones de fase 2, que implican la conversión de sustancias liposolubles en derivados hidrosolubles.
- Reacciones de fase 3, que implican la secreción de la sustancia, sus metabolitos o sus conjugados como parte de la bilis.
Los 3 tipos de reacciones pueden estar vinculados, lo que depende de la composición de la sustancia que se va a eliminar. Por ejemplo, muchos reactantes de la fase 1 no son hidrosolubles y, así, deben experimentar una reacción subsecuente de fase 2 para poder eliminarse. Estas reacciones, que se denominan biotransformaciones, son elementos importantes a considerar en el tratamiento farmacológico.
Reacciones de fase 1
Las reacciones de fase 1 generan la modificación de grupos de medicamentos reactivos mediante oxidación, reducción, hidroxilación u otras reacciones químicas. Casi todas las enzimas que metabolizan fármacos se ubican en las membranas lipofílicas del retículo endoplásmico liso de los hepatocitos. Cuando las membranas se degradan y separan en el laboratorio, se reconstituyen en vesículas denominadas microsomas. Las enzimas en estas membranas a menudo se denominan enzimas microsómicas. Las enzimas implicadas en la mayoría de los procesos de oxidación-reducción de fase 1 son productos de una superfamilia de genes que cuenta con casi 300 miembros. Estos genes codifican una serie de isoenzimas microsómicas que constituyen el sistema citocromo (CYP) P450 (el nombre citocromo P450 deriva de las propiedades espectrales [absorción de luz a 450 nm] de las hemoproteínas que participan en los procesos de oxidación-reducción). Los productos genéticos de muchos de los genes CYP se han identificado y seguido hasta el metabolismo de fármacos específicos y a interacciones potenciales entre los medicamentos. Cada familia de genes es responsable de ciertos procesos del metabolismo de fármacos, y cada miembro de la familia toma a su cargo funciones específicas del metabolismo medicamentoso. Por ejemplo, la familia del gen CYP3 contiene una subfamilia A y varios genes numerados 1, 2, 3 y así, sucesivamente. Por ejemplo, la enzima principal para el metabolismo de la eritromicina en el humano es CYP3A4.
Muchos miembros genéticos del sistema CYP pueden sufrir inducción o supresión de su actividad al tiempo que se realiza la tarea de metabolizar fármacos. Por ejemplo, estimulantes como el alcohol y los barbitúricos pueden inducir a ciertos miembros para incrementar la producción de enzimas, acelerar el metabolismo farmacológico y disminuir la acción farmacológica de la sustancia y los fármacos coadministrados que recurren al mismo miembro del sistema CYP. En el caso de losfármacos que se metabolizan para obtener productos intermedios reactivos, la inducción enzimática puede exacerbar la toxicidad tisular mediada por fármacos. En el sistema del citocromo las enzimas también pueden ser inhibidas por fármacos. Por ejemplo, los medicamentos que contienen imidazoles, como la cimetidina (un fármaco bloqueador de los receptores de histamina tipo 2 utilizado para reducir la secreción de ácido gástrico) y el ketoconazol (un antimicótico) inhiben de manera efectiva el metabolismo de la testosterona. Los contaminantes ambientales son asimismo capaces de inducir la actividad de los genes CYP. Por ejemplo, la exposición al benzo[a]pireno, que se identifica en el humo del tabaco, la carne asada con carbón y otros productos orgánicos de la pirólisis, induce a los miembros de la familia CYP y altera las tasas de metabolismo de algunos fármacos.
Reacciones de fase 2
Las reacciones de fase 2, que implican la conversión de derivados liposolubles en sustancias hidrosolubles, pueden seguir a las reacciones de fase 1 o proceder de manera independiente. La conjugación, que catalizan las enzimas del retículo endoplásmico que acoplan al medicamento con algún compuesto endógeno activado para hacerlo más hidrosoluble, es una de las reacciones más comunes de esta fase. Si bien muchos fármacos hidrosolubles y sustancias endógenas se excretan sin cambios en la orina o la bilis, las sustancias liposolubles tienden a acumularse en el organismo, a menos que se conviertan en compuestos menos activos o metabolitos hidrosolubles. En general, los conjugados son más solubles que el compuesto original y carecen de actividad farmacológica. Debido a que los sustratos endógenos que se utilizan para el proceso de conjugación se obtienen a partir de la dieta, la nutrición desempeña un papel crítico en las reacciones de fase 2.
Una vía alternativa para la conjugación que depende del citocromo P450 es importante para la destoxificación de productos reactivos del metabolismo intermedio. Esta vía recurre al tiol, una sustancia con sulfuro denominada glutatión, que se usa para la conjugación de fármacos que forman grupos electrofílicos con potencial dañino. El glutatión se depleta durante el proceso de destoxificación y debe restituirse de manera constante mediante compuestos obtenidos de la dieta o con fármacos que contienen cisteína, como la N-acetilcisteína. La vía del glutatión es central para la destoxificación de distintos compuestos, entre otros, el acetaminofén (paracetamol), un medicamento analgésico de venta sin receta. El metabolismo del acetaminofén implica una reacción de fase 2. Por lo regular, la capacidad de los reactantes de esta fase 2 es mucho mayor que la que se requiere para metabolizar las dosis recomendadas del medicamento. Sin embargo, en situaciones en que las dosis de acetaminofén son excesivas, la capacidad del sistema de fase 2 se excede y el fármaco se transforma en metabolitos tóxicos que pueden inducir necrosis del hígado si se les permite acumularse. En esta situación, la vía del glutatión desempeña un papel fundamental en la destoxificación de estos metabolitos. Dado que las reservas de glutatión se depletan con rapidez, el medicamento N-acetilcisteína, que actúa como un sustituto del glutatión, se utiliza como tratamiento en caso de sobredosis de acetaminofén. El consumo crónico de alcohol disminuye las reservas de glutatión e incrementa el riesgo de toxicidad por acetaminofén.
Reacciones de fase 3
Las reacciones de fase 3 implican la secreción de los medicamentos, los metabolitos de los medicamentos o los conjugados de los fármacos hacia la bilis. Las proteínas del cajón de unión al ATP tienen participación intrincada en este proceso. Un ejemplo lo constituyen las proteínas de resistencia a fármacos múltiples de los tipos 1, 2 y 3, que transportan medicamentos catiónicos y sus conjugados hacia la bilis.
Además de su papel en el metabolismo de los medicamentos y los químicos, el hígado es responsable de la inactivación o la modificación de las hormonas. La insulina y el glucagón se inactivan por medio de proteólisis o desaminación. La tiroxina y la triyodotironina se metabolizanmediante reacciones que implican desyodación. Las hormonas esteroides, como los glucocorticoides, se inactivan primero por medio de una reacción de fase 1, y luego se conjugan por medio de una reacción de fase 2.
Hepatopatía inducida por fármacos
Como órgano principal para el metabolismo de los fármacos y la destoxificación en el organismo, el hígado se encuentra sujeto a un potencial de daño por una enorme variedad de químicos farmacéuticos y ambientales. Muchos de los fármacos de consumo de tratamiento amplio, entre otros los productos «naturales» que se venden sin receta, pueden inducir lesión hepática. En un estudio multicéntrico reciente, el 10% de 300 casos de lesión hepática inducida por fármacos pudo atribuirse al consumo de productos herbolarios.
Distintos factores del hospedero contribuyen a la susceptibilidad a la hepatopatía inducida por fármacos, entre otros, la predisposición genética, las diferencias de edad, la hepatopatía crónica subyacente, la dieta y el consumo de alcohol, así como el consumo de fármacos con interacciones múltiples. En un estudio reciente en que se analizó a 1198 pacientes con insuficiencia hepática aguda, se identificó la lesión hepática inducida por fármacos como causa de la insuficiencia en el 11,1% de los casos. El diagnóstico temprano de la hepatopatía inducida por fármacos es importante, ya que el retiro de la sustancia resulta curativo en la mayoría de los casos.
Los medicamentos y químicos pueden ejercer sus efectos al inducir lesión o muerte en el hepatocito, o por daño hepático colestásico, secundario al daño a las estructuras de drenaje biliar.
Las reacciones medicamentosas pueden ser predecibles a partir de la estructura química de la sustancia y sus metabolitos, o bien impredecibles (idiosincrásicas), es decir, que dependen de características individuales de la persona que recibe el fármaco.
Lesión hepatotóxicas directa
Se sabe que algunos medicamentos tienen efectos tóxicos sobre el hígado; esto, como consecuencia de su estructura química y la forma en que se metabolizan en ese órgano. El daño hepático directo con frecuencia depende de la edad y la dosis. Las reacciones hepatotóxicas directas suelen ser una característica reconocida en ciertos medicamentos. Por lo general, derivan del metabolismo de la sustancia y la generación de metabolitos tóxicos. Dada la mayor actividad de las enzimas para metabolizar los fármacos en las zonas centrales del hígado, éstos a menudo inducen necrosis centrolobulillar. El acetaminofén, los antimicrobianos, los medicamentos antipsicóticos, los hipolipemiantes y los antiinflamatorios no esteroideos (AINE) son los que la mayoría de las veces se asocian a la lesión hepática aguda. La toxicidad por acetaminofén se caracteriza por elevaciones marcadas de los valores de ATL y ATS, con elevación mínima de la fosfatasa alcalina. Las concentraciones de bilirrubina muestran incremento en forma invariable, y el pronóstico a menudo es peor cuando la necrosis hepatocelular está acompañada por ictericia.
Reacciones idiosincrásicas
En contraste con las reacciones medicamentosas hepatotóxicas directas, las reacciones por idiosincrasia son impredecibles, no se relacionan con la dosis y en ocasiones se acompañan de características que sugieren una reacción alérgica. En algunos casos, la reacción deriva de manera directa de un metabolito que se produce sólo en determinadas personas, según una predisposición genética. Por ejemplo, ciertos individuos tienen capacidad de acetilar con rapidez la isoniazida, un fármaco antifímico.
Reacciones colestásicas
Las reacciones medicamentosas colestásicas dan origen a una disminución en la secreción de la bilis o a la obstrucción del árbol biliar. La colestasis intrahepática aguda es uno de los tipos más frecuentes de reacción idiosincrásica medicamentosa. Entre los fármacos en que se reconoce la inducción de estas reacciones se encuentran el estradiol; la clorpromazina, un antipsicótico, y algunos antibióticos, como amoxicilina/ácido clavulánico, eritromicina y nafcilina.
En forma característica, las reacciones medicamentosas colestásicas se distinguen por un desarrollo temprano de ictericia y prurito, con alteraciones escasas en la sensación de bienestar general de la persona. Los síntomas de colestasis aguda inducida por fármacos ceden una vez que la sustancia se retira, pero la función secretora biliar se recupera a una velocidad más lenta que la que se observa para la función del hígado mismo.
Hepatitis crónica
Algunos fármacos inducen una variante más indolente de daño hepático que tiene similitud estrecha con la hepatitis autoinmunitaria. La identificación temprana de la hepatitis crónica relacionada con medicamentos suele ser difícil. La cirrosis puede desarrollarse antes de que se diagnostique la hepatitis. Reconocer el medicamento responsable del daño hepático puede ser complejo en forma retrospectiva si la persona ha estado consumiendo alcohol o utilizando distintos fármacos.
Hepatitis viral
La hepatitis hace referencia a la inflamación del hígado. Puede ser causada por virus hepatotrópicos que afectan de manera primordial las células hepáticas o hepatocitos, por mecanismos autoinmunitarios o reacciones por fármacos y toxinas, o bien ser secundaria a otros trastornos sitstémicos. Los virus que inducen enfermedad sistémica y pueden afectar al hígado incluyen al virus Epstein-Barr (mononucleosis infecciosa), capaz de generar hepatitis leve durante la fase aguda; el citomegalovirus, en particular en neonatos y personas con inmunosupresión; los herpesvirus, y los enterovirus.
Los virus hepatotrópicos conocidos incluyen al virus de la hepatitis A (VHA), al virus de la hepatitis B (VHB), al virus δ asociado con el virus de la hepatitis B (VHD), al virus de la hepatitis C (VHC) y al virus de la hepatitis E (VHE). Si bien todos éstos pueden inducir hepatitis aguda, difieren en cuanto al modo de transmisión y período de incubación; el mecanismo, grado y cronicidad del daño hepático, y la capacidad para evolucionar al estado de portador. La presencia de antígenos virales y sus anticuerpos puede determinarse mediante pruebas de laboratorio. Los estudiosepidemiológicos han indicado que algunos casos de hepatitis infecciosa se deben a otros factores. Un agente viral similar al VHC se clonó e identificó como virus de la hepatitis G (VHG), también denominado GBV-C. Se ha identificado evidencia de VHG en el 2% de los donadores de sangre en Estados Unidos. Sin embargo, el VHG no se vincula con alguna hepatopatía o con exacerbaciones de la enfermedad hepática.
Etiología y patogénesis
Existen 2 mecanismos de lesión hepática en la hepatitis viral: la lesión celular directa y la inducción de respuestas inmunitarias contra los antígenos virales. Los mecanismos de la lesión han sido estudiados en forma más detallada para el VHB. Se piensa que el grado de inflamación y necrosis depende de la respuesta inmunitaria de la persona. En concordancia, podría esperarse que una respuesta inmunitaria rápida durante la fase aguda de la infección indujera lesión celular, pero que al mismo tiempo eliminara al virus. Así, los pacientes que responden con menos síntomas y con una respuesta inmunitaria marginal tienen menos probabilidad de eliminar al virus, por lo que los hepatocitos que expresan a los antígenos virales persisten, lo que conduce a un estado de cronicidad o de portación crónica. La hepatitis fulminante puede explicarse desde la perspectiva de una respuesta inmunitaria acelerada con necrosis hepática intensa.
La evolución clínica de la hepatitis viral determina distintos síndromes, lo que incluye la infección asintomática con sólo evidencia serológica del trastorno, la hepatitis aguda, el estado de portador sin enfermedad clínica aparente o con hepatitis crónica, la hepatitis crónica con o sin evolución a cirrosis, o a la enfermedad fulminante con desarrollo temprano de insuficiencia hepática. No todos los virus hepatotrópicos provocan cada uno de los síndromes clínicos.
Manifestaciones clínicas
Las manifestaciones agudas de la hepatitis viral pueden dividirse en 3 fases: el período prodrómico o preictérico, el período ictérico y el período de recuperación. Las manifestaciones del primer período varían de lo abrupto a lo insidioso, con malestar general, mialgias, artralgias, tendencia a la fatiga y anorexia. También pueden presentarse síntomas gastrointestinales, como náuseas, vómito y diarrea o constipación. Las concentraciones séricas de ATS y ATL muestran incrementos variables durante el período preictérico de la hepatitis aguda y preceden la elevación de la bilirrubina que acompaña al inicio de la fase ictérica o de ictericia de la infección. En caso de presentarse, la fase ictérica suele seguir a la fase prodrómica, entre 7 y 14 días después. Las personas desarrollan hipersensibilidad a la palpación en torno al área del hígado, pérdida ponderal leve y angiomas en araña. La ictericia tiene menos probabilidad de presentarse en la infección por VHC.
La fase de recuperación se caracteriza por un incremento en la sensación de bienestar, la recuperación del apetito y la resolución de la ictericia. La enfermedad aguda suele ceder de manera gradual en un período de 2 a 12 semanas, con una recuperación clínica completa en un lapso que va de 1 a 4 meses, según el tipo de hepatitis. La infección por VHB y VHC puede inducir un estado de portador, en que la persona no presenta síntomas pero alberga al virus y puede, por ende, transmitir la enfermedad. También hay evidencia que indica que existe un estado de portador para la infección por VHD, pero no para la infección por VHA. Existen 2 tipos de portadores: los sanos, que tienen escasos efectos o carecen de ellos, y los que presentan enfermedad crónica, que pueden o no presentar síntomas.
Los factores que incrementan el riesgo de desarrollar el estado de portador son la edad en el momento de la infección y la condición inmunitaria. Para las infecciones que se desarrollan en una fase temprana de la vida, como en los neonatos con madres con infección por VHB, el estado de portador puede presentarse hasta en el 90% de los casos. Otras personas con riesgo elevado de convertirse en portadores sonlas que presentan anomalías de la inmunidad, las que han recibido transfusiones o hemoderivados en ocasiones múltiples, quienes se encuentran en hemodiálisis y las personas con adicción a drogas.
Hepatitis A
La hepatitis A se debe al VHA, un virus pequeño y sin cubierta que cuenta con una sola cadena de ácido ribonucleico (ARN). Por lo general es una enfermedad benigna y autolimitada; no obstante, puede inducir hepatitis fulminante aguda y muerte, o hacer necesario el trasplante en el 0,15% al 0,2% de los casos.
Etiología y patogénesis
La hepatitis A se contrae de manera primordial por vía orofecal. Cuenta con un período breve de incubación, de 14 a 28 días. El virus se multiplica en el hígado y se excreta en la bilis, para eliminarse en las heces. La eliminación fecal del VHA tiene lugar durante las 2 primeras semanas de la enfermedad. Ésta se presenta a menudo de manera esporádica o en epidemias.
El consumo de leche o agua contaminadas, así como de moluscos obtenidos de aguas infectadas, son vías bastante comunes para la transmisión. Las personas que viajan a otros países y no se han expuesto antes al virus corren un riesgo especial. Debido a que los niños pequeños se mantienen asintomáticos, desempeñan un papel importante en la diseminación de la enfermedad. Las instituciones que albergan a gran número de personas (por lo general, niños) a veces son afectadas por alguna epidemia de hepatitis A.
La conducta oral y la falta de entrenamiento para las evacuaciones favorecen la infección viral entre niños que acuden a guarderías preescolares, que luego llevan el virus a casa, hasta sus hermanos mayores y progenitores. Por lo regular, la hepatitis A no se transmite mediante la transfusión de sangre o de derivados del plasma, lo que, se presume, se debe a que su período corto de viremia suele coincidir con la enfermedad clínica, así que el trastorno resulta aparente y no se aceptan las donaciones de sangre.
Manifestaciones clínicas
El inicio de los síntomas suele ser abrupto, e incluir fiebre, malestar general, náuseas, anorexia, malestar abdominal, coluria e ictericia. Los síntomas de presentación varían según la edad, y la gravedad de éstos aumenta en los grupos de mayor edad. Los niños menores de 6 años con frecuencia son asintomáticos y pocos desarrollan ictericia. La enfermedad en niños mayores y adultos es por lo regular sintomática, y la ictericia se desarrolla en alrededor del 70% de los casos. En general, los síntomas persisten durante alrededor de 2 meses, pero pueden durar más. El VHA no genera hepatitis crónica ni induce un estado de portador.
Marcadores serológicos
Los anticuerpos contra el VHA (anti-VHA) aparecen en una fase temprana de la enfermedad y tienden a persistir en el suero (figura 46-9).
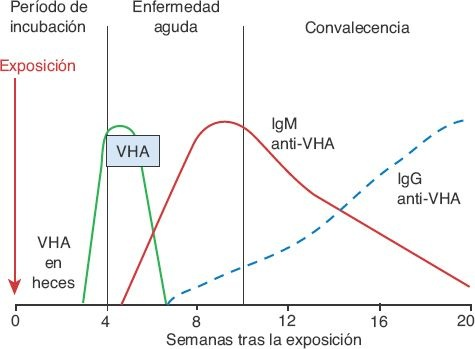
Los anticuerpos tipo inmunoglobulina (Ig) M suelen presentarse durante la primera semana de la enfermedad sintomática y disminuyen con lentitud en un período de 3 a 4 meses. Su presencia coincide con una disminución de la eliminación fecal del virus. Las concentraciones máximas de IgG se desarrollan después de un mes de iniciada la enfermedad y pueden persistir durante toda la vida; aportan inmunidad protectora a largo plazo contra la reinfección. La IgM anti-VHA revela la presencia de hepatitis A aguda, en tanto que la IgG anti-VHA sólo permite documentar una infección previa.
Inmunización
Se dispone de una vacuna contra el virus de hepatitis A. La inmunización pretende sustituir el consumo de inmunoglobulina en personas con riesgo elevado de exposición al VHA; entre otras, quienes viajan a otros países, a regiones en donde el saneamiento es deficiente y existen tasas altas de infección endémica por VHA; los niños que viven en comunidades con tasas altas de infección por VHA, los varones homosexuales activos y los usuarios de drogas ilícitas.
Las personas con hepatopatía crónica preexistente también pudieran beneficiarse con la inmunización. La vacunación de quienes muestran un aumento en el potencial de transmitir la enfermedad (ej. manipuladores de alimentos) también pudiera derivar en beneficio para la salud pública. Los Centers for Disease Control and Prevention (CDC) recomendaron en fecha reciente la vacunación de los niños en los estados, condados y comunidades con tasas altas de infección. Dado que la vacuna aporta poco beneficio para la prevención de la hepatitis en personas con exposición conocida al VHA, en ellas se recomienda la administración de IgG.
Hepatitis B
La hepatitis B se debe al VHB, un virus de ácido desoxirribonucleico (ADN) de doble cadena. El virión completo, también conocido como partícula Dane, está constituido por una cubierta externa y una nucleocápside interna que contiene el ADN del VHB y la polimerasa del ADN (figura 46-10).
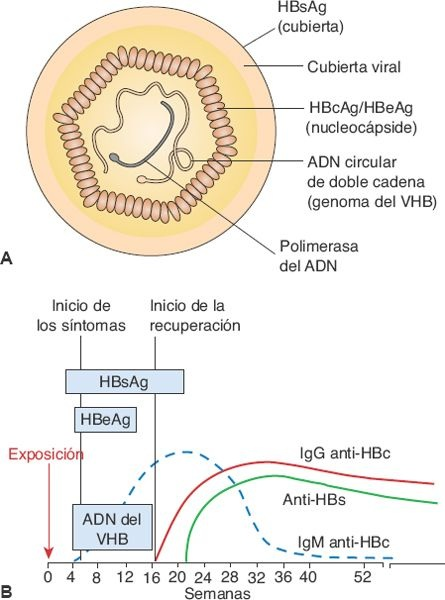
La infección por VHB puede inducir hepatitis aguda, hepatitis crónica, evolución de hepatitis crónica a cirrosis, hepatitis fulminante con necrosis hepática masiva y estado de portador. También participa en el desarrollo de la hepatitis D (hepatitis δ).
En todo el mundo, 350 millones de personas padecen infecciones a largo plazo por el VHB. En Estados Unidos, la incidencia de hepatitis B aguda disminuyó en un 82% desde 1991, debido a una iniciativa nacional. En 2006, la incidencia general (1,6 casos por 100.000 personas) fue la más baja registrada hasta el momento, y representa un descenso del 81% desde que en 1991 se implementara la estrategia de vacunación infantil a nivel nacional. Si bien la incidencia ha disminuido en personas de 25 a 44 años, las tasas para este grupo de edad, en particular en varones, sigue siendo sustancialmente superior que para otros grupos de edad, lo que señala la necesidad de contar con programas de vacunación que tengan como blanco las poblaciones de alto riesgo.
La hepatitis B tiene un período de incubación mayor y representa un problema de salud más grave que la hepatitis A. El virus suele transmitirse por medio de la inoculación con sangre o suero infectados. Sin embargo, el antígeno viral puede identificarse en casi todas las secreciones corporales y diseminarse mediante contacto oral o sexual. En Estados Unidos, la mayoría de las personas con hepatitis B adquiere la infección durante la edad adulta o la adolescencia.
El trastorno tiene gran prevalencia entre los usuarios de drogas intravenosas, los heterosexuales con múltiples pa rejas sexuales, y los varones que mantienen relaciones sexuales con varones. Los trabajadores de la atención de la salud se encuentran en riesgo debido a la exposición a la sangre y las lesiones accidentales con agujas. Si bien el virus puede diseminarse por medio de la transfusión o la administración de he moderivados, las técnicas de rutina para detección han reducido en grado significativo la transmisión por esta ruta. El riesgo de hepatitis B en los neonatos cuyas madres cursan con infección por VHB varía entre el 10% y el 85%, lo que depende de la condición de la madre en relación con el virus. Los lactantes que desarrollan infección tienen un riesgo del 90% de convertirse en portadores crónicos, y hasta el 25% muere por hepatopatía crónica durante la edad adulta.
Marcadores serológicos
Tres antígenos bien identificados se relacionan con el virus: un antígeno nuclear, el HBcAg, que se ubica en la nucleocápside; un transcrito polipeptídico más largo con regiones prenucleares y nucleares, que se designa HBeAg; y un antígeno de superficie, el HBsAg, que se localiza en la cubierta externa del virus. La región prenuclear dirige al polipéptido HBeAg hacia la sangre, en tanto que el HBcAG permanece en los hepatocitos para controlar el ensamblaje de los viriones nuevos.
Los antígenos del VHB evocan la síntesis de anticuerpos específicos: los anti-HBs, los anti-HBcy los anti-HBe. Estos antígenos (el HBcAg no circula libre en la sangre) y sus anticuerpos sirven como marcadores serológicos para dar seguimiento a la evolución de la enfermedad (figura 46-10). El HBsAg es el antígeno viral que, como rutina, se cuantifica en la sangre la mayoría de las veces. Aparece antes del desarrollo de los síntomas, alcanza un valor máximo durante la enfermedad clínica y luego disminuye hasta alcanzar niveles indetectables en 3 a 6 meses. Su persistencia después de los 6 meses señala una multiplicación viral persistente, la infectividad y el riesgo de desarrollar hepatitis crónica.
El HBeAg aparece en el suero poco después del HBsAg, e implica la presencia de multiplicación viral activa. La IgM anti-HBc se vuelve detectable poco después del inicio de los síntomas, en forma concurrente al inicio de la elevación de las transaminasas séricas. En el transcurso de los meses, el anticuerpo tipo IgM es sustituido por IgG anti-HBc. El anti-HBe se vuelve detectable poco después de la desaparición del HBeAg, y su aparición señala el inicio de la resolución de la enfermedad aguda. La IgG anti-HBs, un anticuerpo específico contra el HBsAg, aparece en casi todas las personas una vez que se elimina este último. El desarrollo de anti-HBs señala la recuperación de la infección por VHB, la carencia de infectividad y la protección contra la infección de VHB en el futuro. El anti-HBs es el anticuerpo que se encuentra en personas que tuvieron una inmunización exitosa contra el VHB.
La presencia de ADN viral (ADN del VHB) en el suero es el indicador más certero de hepatitis B. Tiene presencia transitoria durante el período presintomático y por un período breve durante la enfermedad aguda. La presencia de la polimerasa del ADN, la enzima que sirve para la multiplicación del virus, suele ser transitoria, pero persiste durante años en individuos que son portadores crónicos y es un indicador de que la infectividad persiste.
Inmunización
La vacuna contra la hepatitis B aporta protección a largo plazo (hasta 20 años en algunos casos) contra la infección por VHB. El HBsAg es el antígeno que se utiliza para las vacunas contra la hepatitis B. Las vacunas disponibles en Estados Unidos recurren a tecnología de ADN recombinante para expresar el HBsAg en una levadura, que luego se purifica mediante técnicas bioquímicas y biofísicas. La vacuna se encuentra disponible como una formulación que contiene un solo antígeno y también en una combinación fija con otras vacunas. Se dispone de inmunoglobulina contra la hepatitis B (HBIg). Preparada a partir de donadores de plasma con concentraciones elevadas de anti-HBs, se utiliza como adyuvante a la vacuna de la hepatitis B para la inmunoprofilaxia posexposición y así prevenir la infección por VHB en poblaciones de alto riesgo.
Los CDC recomiendan la vacunación de todo los niños de 0 a 18 años de edad como medio para prevenir la transmisión del VHB. También se sugiere para los adultos que no han sido vacunados y se encuentran en una de las siguientes categorías:
- Personas con riesgo elevado de infección por exposición sexual, lo que incluye a las parejas sexuales de personas positivas a HBsAg, individuos con actividad sexual que no se encuentran en una relación monógama mutua a largo plazo, personas que solicitan valoración para tratar enfermedades de transmisión sexual, y varones que tienen relaciones sexuales con varones.
- Personas con riesgo elevado de infección por exposición percutánea o mucosa a la sangre, lo que incluye a usuarios activos y recientes de drogas inyectables, contactos casuales con personas con positividad HBsAg, residentes y personal de instituciones para personas con discapacidad del desarrollo, trabajadores de la atención de la salud y de la seguridad pública con un riesgo anticipado razonable de exposición a la sangre o a fluidos corporales contaminados por sangre, y personas con nefropatía crónica (prediálisis, hemodiálisis, diálisis peritoneal y pacientes que se dializan en casa).
- Otros individuos, lo que incluye a quienes viajan fuera de Estados Unidos a regiones conniveles altos e intermedios de infección endémica por VHB, personas con hepatopatía crónica, con infección por el virus del inmunodeficiencia humana (VIH), y todas las personas que solicitan protección contra la infección por VHB. Los CDC también recomiendan que las mujeres embarazadas se sometan, por rutina, a la detección del HBsAg durante una consulta prenatal temprana, y que los neonatos con madres con HBsAg positivo reciban dosis apropiadas de HBIg y la vacuna contra la hepatitis B.
La HBIg puede ser efectiva en personas que no están vacunadas y se encuentran expuestas a la infección, siempre y cuando se administre en el transcurso de 7 días después de la exposición. La vacunación contra la hepatitis se recomienda para la profilaxis preexposición y posexposición.
Hepatitis C
El VHC es la etiología más frecuente de la hepatitis crónica, la cirrosis y el cáncer hepatocelular en el mundo. Alrededor de 3,2 a 5 millones de estadounidenses se encuentran infectados por VHC. Antes de 1990, la principal vía de transmisión del VHC eran las transfusiones sanguíneas o los hemoderivados contaminados. Con la implementación de las pruebas para el VHC en los bancos de sangre, el riesgo de infección por el virus derivado de una transfusión hemática casi se eliminó en Estados Unidos y en otros países desarrollados. Sin embargo, los procedimientos médicos inseguros y las transfusiones hemáticas realizadas sin detección pueden ser las fuentes más importantes de infección por VHC en países menos desarrollados del mundo.
En la actualidad, el consumo recreacional de drogas inyectables es la modalidad más frecuente de transmisión del VHC en Estados Unidos y Canadá. Las conductas sexuales de alto riesgo, que se definen como la práctica de relaciones sexuales con parejas múltiples o aquellas que se mantienen con una pareja con infección por VHC, ocupan en la actualidad el segundo lugar como factor de riesgo en Estados Unidos. La tasa de transmisión a neonatos nacidos de madres positivas al ARN del VHC varía del 4,6% al 10%. El VHC también puede diseminarse mediante exposición en el ámbito de la atención de la salud, de manera primordial por lesiones por punción con aguja. Asimismo, existe inquietud sobre la posibilidad de que la transmisión de pequeñas cantidades de sangre durante la realización de tatuajes, la acupuntura y el perforado corporal pudiera facilitar la transmisión del VHC.
El VHC es un virus de ARN monocatenario, con propiedades similares a las de los flavivirus, un género de la familia Flaviviridae que incluye los virus de la fiebre amarilla y de la encefalitis de San Luis. Su genoma contiene un marco de lectura abierto único que codifica una poliproteína con alrededor de 3.000 aminoácidos. El producto de la transcripción se escinde en proteínas independientes, lo que incluye a 3 de tipo estructural (una proteína de la cápside y 2 de la cubierta) y 4 de tipo no estructural. El virus presenta inestabilidad genética, lo que conduce a la presencia de genotipos y subtipos múltiples. Se han reconocido 6 genotipos distintos y más de 70 subtipos del virus. Los genotipos 1a y b generan la mayor parte de las infecciones en Estados Unidos. Es probable que la diversidad amplia de genotipos contribuya a la patogenicidad del virus, al permitirle escapar de las acciones de los mecanismos inmunitarios del hospedero y a los fármacos antivirales, al igual que a las dificultades para el desarrollo de una vacuna profiláctica. El desarrollo de una vacuna y de medidas terapéuticas también se ha visto impedido ante la carencia de un sistema de cultivo confiable, reproducible y eficiente para la propagación del virus.
Manifestaciones clínicas
El período de incubación del VHC varía entre 2 y 26 semanas (promedio, 6 a 12 semanas). Casi todos los niños y adultos que adquieren la infección suelen mantenerse sintomáticos. La ictericia es rara y sólo el 10% de los adultos sintomáticos la desarrolla. Estossíntomas suelen durar entre 2 y 12 semanas. La insuficiencia hepática fulminante es rara, y sólo existen algunos casos informados. Una minoría de personas con infección reciente por VHC elimina la infección, pero casi todas (del 85% al 90%) desarrollan hepatitis crónica. Entre los factores asociados con la eliminación espontánea de la infección por VHC parecen encontrarse la edad menor, el sexo femenino y ciertos genes de histocompatibilidad. Las complicaciones más graves de la infección crónica por VHC son la fibrosis hepática progresiva que conduce al desarrollo de cirrosis, la hepatopatía en fase terminal y el cáncer hepatocelular. Los factores del hospedero que pudieran exacerbar el avance de la hepatopatía incluyen la edad mayor al inicio de la infección, el sexo masculino, un estado de inmunosupresión, la infección concurrente por VHB, el consumo de alcohol y el consumo de fármacos hepatotóxicos.
Marcadores serológicos
Se dispone de pruebas de anticuerpos y virales para detectar la presencia de infección por VHC (figura 46-11). Pueden obtenerse resultados negativos falsos en personas con inmunocompromiso y en una fase temprana de la evolución de la enfermedad, antes de que se desarrollen anticuerpos.
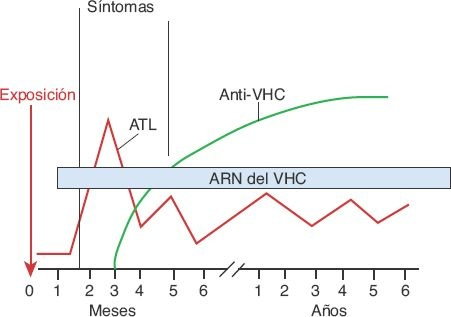
La cuantificación directa del VHC en el suero sigue siendo la prueba más precisa para identificar la infección. Las pruebas virales son muy sensibles y específicas, pero más costosas que las de anticuerpos. Con las técnicas más recientes para análisis de anticuerpos, a menudo la infección puede detectarse incluso entre 6 y 8 semanas después de la exposición, o bien en 1 o 2 semanas con las pruebas virales que recurren a las técnicas de polimerasa de reacción en cadena. A diferencia de la hepatitis B, los anticuerpos contra el VHC no son protectores, pero sirven como marcadores de la enfermedad.
Hepatitis D y E
El virus de la hepatitis D, de la familia Deltaviridae, es el único virus de ARN del género. Es incompleto, en el sentido de que requiere la asistencia del VHB para poder multiplicarse. Puede inducir hepatitis aguda o crónica. La infección depende de la presencia de infección concurrente por VHB; de manera específica, la presencia de HBsAg. La hepatitis D aguda tiene 2 variantes: la coinfección primaria, que se presenta de manera simultánea a un cuadro de hepatitis B aguda, y la sobreinfección, en la que la hepatitis D se sobrepone a una hepatitis B crónica. El factor δ con frecuencia incrementa la gravedad de la infección por VHB. Puede convertir una infección leve por VHB en una hepatitis fulminante grave, inducir hepatitis aguda en portadores asintomáticos o incrementar la tendencia de evolución a la hepatitis crónica y la cirrosis.
Las vías de transmisión de la hepatitis D son similares a las de la hepatitis B. En Estados Unidos, la infección se restringe en gran medida a personas con riesgo elevado de presentar infección por VHB, en específico los usuarios de drogas inyectables. El riesgo más alto se identifica en portadores de VHB. Estas personas deben ser informadas sobre los riesgos de la sobreinfección por VHD.
La hepatitis D se diagnostica mediante la detección de anticuerpos contra VHD (anti-VHD) en el suero o de ARN de VHD en el suero. Se carece de tratamiento específico para la enfermedad. Dado que la infección se vincula con la hepatitis B, la infección por hepatitis D debe iniciar con la prevención de la hepatitis B mediante la vacunación.
El VHE es un virus de ARN de una sola cadena que carece de cubierta. Se transmite por vía orofecal y genera manifestaciones de hepatitis aguda similares a las de la hepatitis A. El virus con el genotipo 3 se ha vinculado con la infección crónica por VHE. Los receptores de trasplante de órgano sólido, la infección por VIH, la quimioterapia y las condiciones hematológicas se han identificado junto con variantes crónicas de la enfermedad. La característica distintiva del VHE es su tasa de mortalidad elevada. En regiones como el sur de Asia, donde la enfermedad muestra prevalencia considerable, la tasa de mortalidad en mujeres embarazadas es del 51%, debido al desarrollo de hepatitis fulminante. En Estados Unidos, los casos informados afectan a personas que en fecha reciente visitaron un área endémica. Los individuos sin antecedente de viaje se afectan en casos muy poco frecuentes.
Hepatitis viral crónica
La hepatitis crónica se define como una reacción inflamatoria crónica del hígado con más de 3 a 6 meses de duración. Se determina por la persistencia de concentraciones elevadas de aminotransferasas séricas y hallazgos histológicos característicos en la biopsia hepática. Las características clínicas de la hepatitis viral crónica son muy variables y no predicen la evolución. Los síntomas más frecuentes son fatiga, malestar general, pérdida del apetito y episodios ocasionales de ictericia. La elevación en las concentraciones de las aminotransferasas en el suero depende del nivel de actividad de la enfermedad.
La hepatitis viral crónica es la causa principal de hepatopatía crónica, cirrosis y cáncer hepatocelular en el mundo, y en la actualidad ocupa el primer lugar entre las causas que obligan a recurrir al trasplante hepático en el adulto. Entre los virus hepatotrópicos, sólo 3 se reconocen como causantes de hepatitis crónica, VHB, VHC y VHD. La hepatitis B, que en comparación con la hepatitis C tiene menos probabilidad de convertirse en una infección crónica, genera entre el 5% y el 10% de los casos de hepatopatía crónica y cirrosis en Estados Unidos. Se caracteriza por la persistencia del ADN del VHB y, por lo general, del HBeAg en el suero, que señalan la multiplicación viral activa. Muchas personas se encuentran asintomáticas en el momento del diagnóstico, y el primer signo de infección es la elevación en las concentraciones de aminotransferasas en el suero. La infección crónica por hepatitis D depende de la infección concurrente por VHB.
La hepatitis B crónica genera la mayoría de los casos de hepatitis viral crónica. La infección por VHC se vuelve crónica en el 60% al 85% de los pacientes. La infección crónica por VHC con frecuencia mantiene una intensidad baja durante varios años, mientras destruye de manera silenciosa las células hepáticas. Casi todas las personas con hepatitis C crónica se encuentran asintomáticas, y el diagnóstico suele establecerse tras la detección de concentraciones elevadas de aminotransferasas en el suero, o por referencias de fatiga o debilidad inespecíficas. Debido a que la evolución de la hepatitis C aguda es a menudo leve, muchas personas no recuerdan los episodios de la infección aguda.
Tratamiento
No existen estrategias terapéuicas simples y efectivas para la hepatitis viral crónica.
Los medicamentos que se utilizan para el tratamiento de la infección crónica por VHB incluyen los interferones (2α-interferón recombinante y peginterferón) y los fármacos antirretrovirales análogos nucleótidos y nucleósidos (lamivudina, entecavir y tenofovir). Las personas con multiplicación viral activa pueden recibir tratamiento con interferón (α-2a-interferón pegilado).
Los peginterferones tienen una semivida prolongada en el suero y se administran una vez a la semana. El 50% de las personas tratadas responde con una normalización sostenida en las concentraciones de enzimas hepáticas, la desaparición del HBeAg y el ADN del VHB del suero, la aparición de anti-HBe y una mejoría en la sobrevivencia. Los análogos nucleósidos y nucleótidos pueden utilizarse como alternativa al interferón para el tratamiento de la infección crónica por VHB y se toleran mejor.
La lamivudina puede administrarse por vía oral y suele ser bien tolerada; no obstante, se relaciona con una tasa más alta de resistencia viral, una tasa más baja de respuesta duradera y una mayor necesidad de tratamiento prolongado, en comparación con el interferón.
El entecavir, otro análogo nucleósido, puede usarse para el tratamiento de personas con resistencia a la lamivudina o cirrosis.
El tenofovir, un medicamento empleado para el tratamiento de la infección por VIH, también tiene actividad considerable contra el VHB.
Otros medicamentos antivirales se encuentran en estudio y es probable que se investiguen estrategias que recurren a fármacos múltiples.
En personas con hepatitis D concurente, el tratamiento con interferón puede inducir la normalización en las concentraciones de aminotransferasas, un mejoramiento histológico y la eliminación del ARN del VHD del suero en cerca del 50% de los casos, pero es frecuente la recaída una vez que se suspende el tratamiento. La lamivudina no es efectiva en la hepatitis D crónica.
El tratamiento actual para las personas que no han sido tratadas por hepatitis C crónica es una combinación de las nuevas variantes pegiladas del interferón (2b-α o 2a-α) combinado con ribavirina (un análogo nucleósido).
El tratamiento con peginterferón y ribavirina es costoso y sus efectos colaterales, que incluyen síntomas similares a la influenza, son casi universales. Los efectos colaterales más graves, entre otros los síntomas psiquiátricos (depresión), la disfunción tiroidea y la depresión de la médula ósea, son menos frecuentes. Si bien casi todas las personas con infección por VHC son candidatas para el tratamiento, muchas tienen otros problemas de salud que constituyen contraindicaciones a éste.
El trasplante hepático es una opción terapéutica para la hepatopatía en fase terminal debida a la hepatitis viral. Ha tenido más éxito en individuos con hepatitis C que en aquellos que padecen hepatitis B. Si bien es frecuente que el injerto se reinfecte, la enfermedad parece evolucionar con más lentitud.
Hepatitis autoinmunitaria
Es una variante grave de hepatitis crónica de origen desconocido; se relaciona con hepatitis de interfase, autoanticuerpos circulantes e hipergammaglobulinemia. Si bien el trastorno suele identificarse en mujeres jóvenes, puede presentarse en personas de ambos sexos y a cualquier edad.
Las observaciones clínicas y de laboratorio llevaron a la hipótesis de que la hepatitis autoinmunitaria es un trastorno multifactorial, con factores genéticos y ambientales que desempeñan papeles importantes. La mayor parte del conocimiento sobre la genética del trastorno deriva de los genes del antígeno leucocitario humano (ALH) que residen en el complejo mayor de histocompatibilidad (CMH), ubicado en el brazo corto del cromosoma 6. Los factores ambientalesque, se asume, inducen hepatitis autoinmunitaria no se han identificado, pero incluyen virus y químicos.
Existen 2 tipos distintos de hepatitis autoinmunitaria. La hepatitis autoinmunitaria tipo I , la variante más común del trastorno, se caracteriza por un incremento en las concentraciones de autoanticuerpos dirigidos contra el músculo liso y el núcleo. Alrededor del 78% de los casos se presenta en mujeres y el 38% de los afectados padece otros trastornos autoinmunitarios. La susceptibilidad a la hepatitis autoinmunitaria tipo I depende sobre todo del gen HLA-DRB1. La hepatitis autoinmunitaria tipo II se presenta de manera especial en niños de 2 a 14 años y se caracteriza por la presencia de anticuerpos contra los microsomas del hígado y el riñón, y el citosol hepático. Con frecuencia, el trastorno se acompaña de otras enfermedades autoinmunitarias, sobre todo, diabetes mellitus tipo 1, vitiligo y tiroiditis. El componente genético de este tipo de hepatitis autoinmunitaria se encuentra menos bien definido.
Manifestaciones clínicas
Las manifestaciones clínicas del trastorno cubren un espectro que se extiende de la ausencia de síntomas aparentes hasta los signos propios de la hepatopatía inflamatoria o la cirrosis. La exploración física pudiera no revelar anomalías, pero también puede permitir la detección de hepatomegalia, esplenomegalia, ictericia y datos de hepatopatía crónica. En los casos asintomáticos es posible descubrir el trastorno al identificar, en suero, concentraciones anómalas de enzimas durante la práctica de pruebas para vigilancia de rutina.
Diagnóstico y tratamiento
El diagnóstico diferencial incluye las medidas para excluir otras causas de hepatopatía, entre otras, hepatitis B y C. Un hallazgo característico de laboratorio es la elevación marcada de las gammaglobulinas en el suero. La biopsia se utiliza para confirmar el diagnóstico.
El tratamiento de elección para este tipo de hepatitis lo constituyen los corticoesteroides y los fármacos inmunosupresores. Si bien algunas personas permanecen en remisión una vez que se suspende el tratamiento, la mayoría requiere tratamiento para mantenimiento a largo plazo. El trasplante hepático puede ser necesario en individuos resistentes o con intolerancia al tratamiento inmunosupresor, y en quienes se desarrolla hepatopatía en fase terminal.
Trastornos biliares intrahepáticos
Las enfermedades biliares intrahepáticas alteran el flujo de bilis en el hígado, con lo que generan colestasis y cirrosis biliar. Entre las causas de la enfermedad biliar intrahepática se encuentran la cirrosis biliar primaria y la cirrosis biliar secundaria.
Cirrosis biliar primaria
La cirrosis biliar primaria (CBP) es una enfermedad crónica del hígado; se caracteriza por la destrucción autoinmunitaria de los conductos biliares intralobares, e induce colestasis. El trastorno se identifica con más frecuencia en mujeres de 40 a 60 años de edad. Se detectan casos familiares en padres e hijos, y entre hermanos. Sin embargo, a diferencia de otros trastornos autoinmunitarios, hay una relación escasa, en caso de existir, con algún alelo específico del MHC. Además, con la posible excepción de un riesgo más alto referido en cuanto a la presencia de un polimorfismo del gen del receptor de la vitamina D, no existen factores genéticos claros que influyan sobre la enfermedad. Al igual que con otros trastornos autoinmunitarios, los posibles desencadenantes ambientales incluyenelementos infecciosos y químicos.
Manifestaciones clínicas
El trastorno se caracteriza por la cicatrización y la destrucción de inicio insidioso y progresivo del tejido hepático. El hígado aumenta su tamaño y adquiere una tonalidad verdosa debido a la bilis acumulada. Los síntomas más tempranos son el prurito inexplicable, la pérdida ponderal y la fatiga; tras éstos se desarrollan coluria y acolia. La osteoporosis se presenta en el 51% de las mujeres. La ictericia es una manifestación tardía del trastorno, al igual que otros signos de insuficiencia hepática. Las concentraciones séricas de fosfatasa alcalina son altas en personas con cirrosis biliar primaria.
Diagnóstico y tratamiento
El diagnóstico se establece cuando la persona tiene 2 de los 3 signos y síntomas siguientes: destrucción de los conductos biliares y presencia de colangitis no supurativa en la biopsia hepática; colestasis con elevación de la fosfatasa alcalina durante por lo menos 6 meses, y presencia de anticuerpos antimitocondriales en las pruebas en sangre.
El tratamiento es en gran medida sintomático. El ácido ursodesoxicólico (ursodiol), el único fármaco autorizado para el tratamiento de la CBP, incrementa el flujo biliar, disminuye la toxicidad de los contenidos biliares y, se ha probado, reduce la velocidad de deterioro clínico. La colestiramina, un fármaco quelante de ácidos biliares, pudiera resultar benéfica para el manejo del prurito. La colchicina, que actúa para impedir la migración leucocitaria y la fagocitosis, y el metotrexate, un medicamento con propiedades inmunosupresoras, también han generado beneficios en términos del alivio sintomático. Se ha demostrado que los corticoesteroides mejoran la histología hepática y las pruebas de función hepáticas en suero, pero conllevan efectos colaterales graves a largo plazo. El trasplante hepático sigue siendo el único tratamiento para la enfermedad avanzada.
La CBP no reincide tras el trasplante hepático si se implementa una inmunosupresión apropiada.
Cirrosis biliar secundaria
La cirrosis biliar secundaria deriva de la obstrucción prolongada del árbol biliar extrahepático. La causa más frecuente es la colelitiasis. Otras causas son las neoplasias malignas del árbol biliar o la cabeza del páncreas, así como las estenosis del conducto biliar común inducidas por procedimientos quirúrgicos previos. La cirrosis biliar extrahepática pudiera mejorar con procedimientos quirúrgicos diseñados para aliviar la obstrucción.
Hepatopatía inducida por alcohol
El espectro de la hepatopatía alcohólica incluye al hígado graso, la hepatitis alcohólica y la cirrosis.
La mayoría de las muertes por cirrosis alcohólica puede atribuirse a insuficiencia hepática, hemorragia de las varices esofágicas o insuficiencia renal. Se calcula que existen alrededor de 14 millones de alcohólicos en Estados Unidos. Cerca del 10% de los alcohólicos desarrolla cirrosis al persistir en el consumo abundante de alcohol.
Metabolismo del alcohol
El alcohol se absorbe con facilidad a partir del tubo gastrointestinal; es una de las pocas sustancias que puede absorberse a partir del estómago. Como sustancia, el alcohol ocupa un sitio entre un alimento y un fármaco. Aporta calorías, pero no puede degradarse o almacenarse como proteínas, grasas o carbohidratos. El metabolismo del alcohol aporta 7,1 kcal/g como alimento.
Entre el 80% y el 90% del alcohol que bebe una persona se metaboliza en el hígado. El resto se excreta por los pulmones, los riñones y la piel.El metabolismo del alcohol (alcohol etílico o etanol) procede de manera simultánea por 2 vías:
la del sistema de la deshidrogenasa alcohólica (DHasaA), que se ubica en el citoplasma de los hepatocitos, y la del sistema microsómico para oxidación del etanol (MEOS), que se ubica en el retículo endoplásmico. Las vías de la DHasaA y el SMOE producen trastornos metabólicos y tóxicos específicos. Una tercera vía con menor importancia, la vía de la catalasa, que tiene lugar en los peroxisomas, es capaz de degradar el etanol en circunstancias inusuales.
La vía principal para el metabolismo del etanol implica a la DHasaA, una enzima que cataliza la conversión del alcohol en acetaldehído. En la oxidación del alcohol mediada por la DHasaA se obtienen tanto acetaldehído como hidrógeno. El hidrógeno (H+) se transfiere al cofactor dinucleótido de nicotinamida adenina (DNA), que se convierte a su forma reducida (DNAH). El acetaldehído formado pierde de nuevo hidrógeno y se metaboliza en acetato; gran parte de éste se libera al torrente sanguíneo. Como consecuencia, el metabolismo del etanol genera un exceso de DNAH, que, se piensa, contribuye al daño hepático que muchas veces acompaña al consumo excesivo de alcohol.
El DNA también es necesario para muchos otros procesos metabólicos, como el metabolismo del piruvato, el urato y los ácidos grasos. Puesto que el alcohol compite por el consumo del DNA, tiende a alterar otras funciones metabólicas del hígado. El consumo preferencial de DNA para el metabolismo del alcohol puede generar un aumento en la producción y la acumulación de ácido láctico en la sangre. Al reducirse la disponibilidad de DNA, el alcohol también compromete la capacidad del hígado para sintetizar glucosa a partir de aminoácidos y de otros precursores. Es posible el desarrollo de hipoglucemia inducida por alcohol cuando se presenta una ingesta excesiva durante los períodos de disminución de las reservas hepáticas de glucógeno.
La vía del SMOE, que se identifica en el retículo endoplásmico liso, genera acetaldehído y radicales libres. La ingesta prolongada y excesiva de alcohol da origen a la inducción enzimática y al aumento de la actividad del SMOE. Una de las enzimas más importantes del SMOE, miembro del sistema CYP P450, también oxida distintos compuestos adicionales, entre otros, varios fármacos (ej. acetaminofén e isoniazida), toxinas (ej. tetracloruro de carbono y halotano), las vitaminas A y D, y fármacos carcinógenos (ej. aflatoxina y nitrosaminas). El aumento en la actividad de este sistema favorece la susceptibilidad de los individuos que consumen alcohol en exceso a los efectos hepatotóxicos de otras sustancias.
Los productos finales del metabolismo del alcohol (ej. a acetaldehído y radicales libres) son responsables de distintas alteraciones metabólicas capaces de inducir daño hepático. El acetaldehído, por ejemplo, tiene efectos tóxicos múltiples sobre los hepatocitos y la función hepática. La edad y el sexo desempeñan un papel sobre el metabolismo del alcohol y la producción de metabolitos lesivos.
El sistema de la DHasaA se deprime por la acción de la testosterona. Así, las mujeres tienden a producir mayores cantidades de acetaldehído y tienen mayor predisposición al daño hepático inducido por el alcohol que los varones. La edad también parece afectar las capacidades para metabolismo del alcohol del hígado y la resistencia a los efectos hepatotóxicos. Por otra parte, los factores genéticos pueden influir sobre la intensidad de la hepatopatía inducida por alcohol. Existen isoenzimas múltiples de la DHasaA, cuyo polimorfismo genético se está estudiando desde la perspectiva de las implicaciones clínicas potenciales.
Hepatopatía alcohólica
El metabolismo del alcohol conduce al ataque químico contra ciertas membranas del hígado, pero se desconoce si el daño es causado por el acetaldehído o por otros metabolitos. Se sabe que el acetaldehído impide la actividad del sistema de transporte de electrones en las mitocondrias, responsable del metabolismo oxidativo y la generación de ATP; como consecuencia, los hidrogeniones que se generan en las mitocondrias se desvían para la síntesis de lípidos y las cetogénesis. En los hepatocitos se identifican acumulaciones anómalas de estas sustancias (ej. hígado graso), al igual que en la sangre. La unión del acetaldehído a otras moléculas compromete la destoxificación de los radicales libres y la síntesis de proteínas. El acetaldehído también promueve la síntesis de colágeno y la fibrogénesis. La lesiones hepatocelulares tienden a tener mayor prevalencia en el área centrolobulillar que circunda la vena central, sitio en que se concentran las vías para el metabolismo del alcohol. Se trata de la región del lobulillo que tiene la tensión de oxígeno más baja; se cree que la concentración baja de oxígeno en esta área del hígado pudiera contribuir al daño.
La cantidad de alcohol que se requiere para producir hepatopatía crónica varía en gran medida, y depende de la complexión corporal, la edad, el sexo y el origen étnico; no obstante, el valor superior del intervalo se aproxima a 80 g por día durante 10 a 12 años. Esta cantidad de alcohol puede consumirse en 240 ml de whisky de 86° (41% de alcohol), 2 botellas de vino o 6 envases de cerveza de 360 ml cada uno. Incluso una vez que se suspende el consumo de alcohol y éste se ha metabolizado, los procesos que dañan a los hepatocitos pueden continuar durante muchas semanas y meses. Los efectos clínicos y químicos con frecuencia empeoran antes de que la enfermedad se resuelva. La acumulación de grasa suele desaparecer en el transcurso de algunas semanas, y la colestasis y la inflamación también ceden al pasar el tiempo. Sin embargo, la fibrosis y la cicatrización persisten. Los lobulillos hepáticos se distorsionan al tiempo que se producen células hepáticas nuevas y generan nódulos.
Si bien el mecanismo por el que el alcohol ejerce sus efectos tóxicos en las estructuras hepáticas es de alguna manera incierto, los cambios que se desarrollan pueden dividirse en 3 fases: cambios grasos, hepatitis alcohólica y cirrosis.
El hígado graso se caracteriza por la acumulación de grasa en los hepatocitos, condición que se denomina esteatosis (figura 46-12).
El hígado adquiere un color amarillo y aumenta su volumen debido a la acumulación excesiva de grasa. La patogénesis del hígado graso no se comprende del todo y pudiera variar según la cantidad de alcohol consumida, el contenido de grasas en la dieta, las reservas corporales de grasa, la condición hormonal y otros factores. Existe evidencia de que la ingestión de grandes cantidades de alcohol es capaz de inducir cambios propios del hígado graso, incluso con una dieta adecuada. Los cambios grasos que tienen lugar con la ingestión de alcohol no suelen generar síntomas y son reversibles una vez que el consumo de la sustancia se suspende.
La hepatitis alcohólica es el estadio intermedio entre los cambios grasos y la cirrosis. Con frecuencia se observa después de un incremento abrupto en el consumo de alcohol y es común en los consumidores de alcohol. Una revisión reciente encontró que tiene una tasa de mortalidad aproximada del 34%. La hepatitis alcohólica se caracteriza por inflamación y necrosis de los hepatocitos. Esta fase suele distinguirse por hipersensibilidad a la palpación hepática, dolor, anorexia, náuseas, fiebre, ictericia, ascitis, insuficiencia hepática; aunque algunas personas pueden mantenerse asintomáticas. La condición siempre es grave y en ocasiones mortal. El pronóstico inmediato se correlaciona con la gravedad de la lesión a la célula hepática. En algunos casos, el trastorno evoluciona con rapidez hacia la insuficiencia hepática y la muerte. En personas que sobreviven y siguen bebiendo, la fase aguda es muchas veces seguida por una hepatitis alcohólica persistente, que evoluciona a cirrosis en cuestión de 1 o 2 años.
La cirrosis alcohólica es el resultado final de los episodios repetidos de lesión hepática relacionada con el consumo de alcohol y señala el inicio de la hepatopatía alcohólica de fase terminal. El aspecto macroscópico del hígado con cirrosis temprana se caracteriza por nódulos finos y uniformes en la superficie del órgano. De manera tradicional, la condición se ha denominado cirrosis micronodular o de Laennec. En la cirrosis más avanzada, los procesos de regeneración hacen que los nódulos crezcan y se vuelvan más irregulares en tamaño y configuración. Al tiempo que esto ocurre, los nódulos hacen que el hígado desarrolle lobulillos nuevos mediante la formación de tractos portales nuevos y canales venosos de flujo de salida. Los nódulos pueden comprimir las venas hepáticas, lo que aleja al flujo sanguíneo del hígado y genera hipertensión portal, desarrollo de cortocircuitos portosistémicos extrahepáticos y colestasis.
Hepatopatía grasa no alcohólica
El término hepatopatía grasa no alcohólica (HGNA) se debe a la disfunción metabólica que afecta al hígado. En Estados Unidos se trata de la variante más frecuente de hepatopatía crónica. La condición puede variar desde la esteatosis simple (infiltración grasa del hígado) hasta la esteatohepatitis no alcohólica (esteatosis con inflamación y necrosis de los hepatocitos). Si bien la esteatosis aislada no parece ser progresiva, alrededor del 10% al 15% de las personas con esteatohepatitis no alcohólica evoluciona a cirrosis. La obesidad, la diabetes mellitus tipo 2, el síndrome metabólico y la hiperlipidemia son factores que a menudo coexisten con la hepatopatía grasa. La enfermedad también se relaciona con otras anomalías nutricionales, condiciones quirúrgicas, fármacos y exposición laboral a toxinas. Tanto la pérdida ponderal rápida como la nutrición parenteral pudieran conducir a la HGNA. El puenteo yeyunoileal, un procedimiento quirúrgico que se utiliza para la pérdida ponderal, se ha abandonado en gran medida por esta causa.
Patogénesis
Se piensa que la patogénesis de la HGNA implica tanto la acumulación de lípidos dentro de los hepatocitos como la formación de radicales libres, de manera similar a lo que tiene lugar con el metabolismo del alcohol. Las anomalías metabólicas principales que conducen a la acumulación de lípidos se conocen poco, pero se piensa que incluyen alteraciones de las vías para la captación, síntesis, degradación y secreción de los lípidos hepáticos, como consecuencia de la resistencia a la insulina. La obesidad incrementa la síntesis de ácidos grasos libres y limita su oxidación. La diabetes mellitus tipo 2 o la resistencia a la insulina también aumentan la lipólisis en el tejido adiposo y la producción subsecuente de ácidos grasos libres. Cuando se rebasa la capacidad del hígado para exportar los triglicéridos, los ácidos grasos excedentes contribuyen al desarrollo del hígado graso.
Tanto las cetonas como los ácidos grasos libres son inductores de las antes descritas enzimas CYP P450 de la vía del SMOE, lo que tiene como consecuencia la formación de radicales libres, entre otros, peróxido de hidrógeno y superóxido.
A continuación tiene lugar una peroxidación anómala de los lípidos; le siguen la lesión directa a los hepatocitos, la liberación de productos colaterales tóxicos, inflamación y fibrosis.
Manifestaciones clínicas
La HGNA suele ser asintomática; no obstante, pueden existir fatiga y malestar en el cuadrante superior derecho del abdomen. Los hallazgos anómalos de laboratorio más comunes y con frecuencia únicos son la elevación leve o moderada en las concentraciones séricas de ATS, ATL o ambas. Otras anomalías, como hipoalbuminemia, prolongación del tiempo de protrombina e hiperbilirrubinemia, pueden presentarse en personas con hepatopatía en fase cirrótica.
Para el diagnóstico de la HGNA se requiere una biopsia hepática y excluir el alcohol como etiología del trastorno.
Tratamiento
El objetivo del tratamiento es reducir la velocidad de progresión de la HGNA y prevenir la enfermedad relacionada con el hígado. Tanto la pérdida ponderal como el ejercicio mejoran la resistencia a la insulina y se recomiendan a la par del tratamiento de los trastornos metabólicos asociados. Debe evitarse el consumo de alcohol. El avance de la enfermedad es lento, y la magnitud de la morbilidad y la mortalidad que se relacionan con ella es incierta. Un estudio reciente demostró que la administración de estatinas y antioxidantes, como vitaminas A y E, ha sido efectiva para reducir el riesgo de desarrollar esteatosis hepática en pacientes con HGNA. El trasplante hepático es una alternativa para algunos individuos con hepatopatía en fase terminal, pero la HGNA puede reincidir hasta en el 39% de los receptores de trasplante hepático.
Cirrosis, hipertensión portal e insuficiencia hepática
Cirrosis
La cirrosis representa la fase terminal de la hepatopatía crónica, en la que gran parte del tejido hepático funcional ha sido sustituido por tejido fibroso. Si bien suele relacionarse con el alcoholismo, puede desarrollarse durante la evolución de otros trastornos, como la hepatitis viral, las reacciones tóxicas por medicamentos y químicos, la obstrucción biliar y la HGNA. La cirrosis también acompaña a trastornos metabólicos que inducen el depósito de minerales en el hígado, como la hemocromatosis (es decir, depósito de hierro) y la enfermedad de Wilson (es decir, depósito de cobre).
La cirrosis se caracteriza por una fibrosis difusa y la conversión de la arquitectura hepática normal en nódulos que contienen hepatocitos en proliferación circundados por fibrosis. La formación de los nódulos, que varían en tamaño desde los muy pequeños (<3 mm, micronódulos) hasta los grandes (varios centímetros, macronódulos), representa un equilibrio entre la actividad de regeneración y la cicatrización constrictiva. El tejido fibroso que reemplaza al tejido hepático con funcionamiento normal forma bandas constrictivas que interrumpen el flujo en los sistemas de canales vasculares y de conductos biliares del hígado. El compromiso de los canales vasculares predispone a la hipertensión portal y sus complicaciones, a la obstrucción de los canales biliares y la exposición a los efectos destructivos de la estasia biliar, así como a la pérdida de los hepatocitos, lo que conduce a la insuficiencia hepática.
Manifestaciones clínicas
Las manifestaciones de la cirrosis son variables, desde la hepatomegalia asintomática hasta la insuficiencia hepática (figura 46-13).
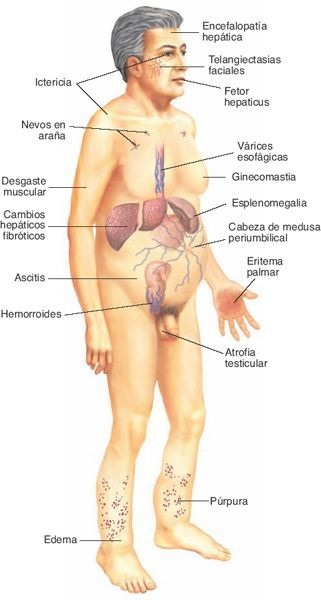
Por lo regular no existen síntomas hasta que el trastorno se encuentra muy avanzado. Los signos y síntomas más frecuentes son la pérdida ponderal (en ocasiones enmascarada por la ascitis), la debilidad y la anorexia. Es frecuente la diarrea, aunque algunas personas pueden referir constipación. La hepatomegalia y la ictericia también son signos comunes de cirrosis. Puede existir dolor abdominal por el crecimiento hepático o la distensión de la cápsula de Glisson. Este dolor se localiza en la región epigástrica o en el cuadrante superior derecho, y se describe como sordo, opresivo y generador de una sensación de plenitud.
Las manifestaciones tardías de la cirrosis se vinculan con la hipertensión portal y la insuficiencia de los hepatocitos. Esplenomegalia, ascitis y cortocircuitos portosistémicos (es decir, várices esofágicas, hemorroides y cabeza de medusa) derivan de la hipertensión portal. Otras complicaciones incluyen la hemorragia por disminución de los factores de la coagulación, la trombocitopenia secundaria a la esplenomegalia, ginecomastia y distribución ginecoide del vello púbico en el varón por efecto de la atrofia testicular, angiomas en araña, eritema palmar y encefalopatía, con asterixis y signos neurológicos.
Hipertensión portal
Se caracteriza por el aumento de la resistencia al flujo en el sistema venoso portal y una presión sostenida en la vena porta. Por lo regular, la sangre venosa que regresa hacia el corazón desde los órganos abdominales se conjunta en la vena porta y viaja por el hígado antes de ingresar a la vena cava. La hipertensión portal puede derivar de distintas condiciones que incrementan la resistencia al flujo sanguíneo hepático, entre otras, obstrucciones prehepáticas, poshepáticas e intrahepáticas (casos en que el término hepático hace referencia a los lobulillos hepáticos y no a todo el hígado). Las causas prehepáticas de hipertensión portal incluyen la trombosis de la vena porta y la compresión externa por cáncer o adenomegalias, que generan obstrucción de la vena porta antes de que ingrese al hígado.La obstrucción poshepática se refiere a cualquier obstrucción al flujo sanguíneo por las venas hepáticas más allá de los lobulillos hepáticos, ya sea dentro del hígado o en algún punto distal. Se debe a condiciones como la trombosis de las venas hepáticas, la enfermedad venooclusiva, y a la insuficiencia cardíaca derecha grave que impide el flujo de salida de la sangre venosa del hígado.
El síndrome de Budd-Chiari alude a la enfermedad hepática congestiva secundaria a la oclusión de venas hepáticas múltiples o a la porción hepática de la vena cava inferior. La causa principal del síndrome de Budd-Chiari es la trombosis de las venas hepáticas, en asociación con distintos trastornos, como la policitemia vera, los estados de hipercoagulabilidad relacionados con los tumores malignos, el embarazo, la infección bacteriana, la enfermedad metastásica hepática y el traumatismo. El síndrome de obstrucción sinusoidal o enfermedad venooclusiva hepática es una variante del síndrome de Budd-Chiari, que la mayoría de las veces se identifica en personas tratadas con ciertos fármacos quimioterapéuticos contra el cáncer, la radiación hepática o el trasplante de médula ósea.
Las etiologías intrahepáticas de hipertensión portal incluyen condiciones que inducen la obstrucción del flujo sanguíneo dentro del hígado. En la cirrosis alcohólica, que es la causa principal de hipertensión portal, bandas de tejido fibroso y nódulos fibrosos distorsionan la arquitectura del hígado y aumentan la resistencia al flujo sanguíneo portal, lo que conduce a la hipertensión portal.
Las complicaciones de la hipertensión portal derivan del aumento de la presión y la dilatación de los canales venosos ubicados en un punto proximal a la obstrucción (figura 46-14). Además, se abren canales colaterales que conectan a la circulación portal con la circulación sistémica.
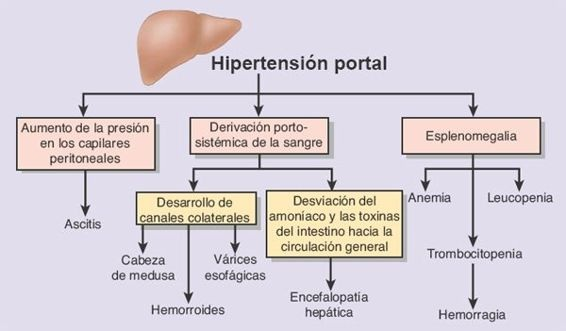
Las complicaciones principales del aumento de la presión en la vena porta y la apertura de los canales colaterales son la ascitis, esplenomegalia, encefalopatía hepática y la formación de cortocircuitos portosistémicos, con hemorragia a partir de las várices esofágicas.
Ascitis
Se presenta cuando se incrementa la cantidad del líquido en la cavidad peritoneal, y constituye una manifestación de fase tardía de la cirrosis y la hipertensión portal.
No es raro que las personas con cirrosis avanzada presenten una acumulación de 15 L o más de líquido ascítico. Estos individuos con frecuencia experimentan malestar abdominal, disnea e insomnio. También pueden cursar con dificultad para caminar o vivir en forma independiente.
Si bien los mecanismos responsables del desarrollo de la ascitis no se comprenden en su totalidad, varios factores parecen contribuir a la acumulación del líquido, entre otros, el aumento de la presión capilar secundario a hipertensión portal y obstrucción al flujo venoso a través del hígado, la retención de sal y agua en el riñón, y la disminución de la presión coloidosmótica debida a la síntesis de albúmina en el hígado. La disminución del volumen sanguíneo (es decir, la teoría del llenado por debajo de lo óptimo) y el volumen sanguíneo excesivo (es decir, la teoría del llenado excesivo) se han utilizado para explicar el incremento de la retención de sal y agua en el riñón. De acuerdo con la teoría del llenado por debajo de lo óptimo, la contracción del volumen sanguíneo efectivo constituye una señal aferente que hace que el riñón retenga sal y agua.
El volumen sanguíneo efectivo puede disminuir debido a la pérdida de líquido hacia el interior de la cavidad peritoneal o como consecuencia de la vasodilatación inducida por la presencia de sustancias vasodilatadoras circulantes. La teoría del llenado excesivo propone que el suceso inicial para el desarrollo de la ascitis es la retención renal de sal y agua generada por trastornos que afectan al hígado mismo. Éstos incluyen la incapacidad del hígado para metabolizar la aldosterona, lo que genera un incremento en la retención de sal y agua en el riñón. Otro factor que podría tiene probabilidad de contribuir a la patogénesis de la ascitis es la disminución de la presión coloidosmótica, que limita la reabsorción del líquido a partir de la cavidad peritoneal.
El tratamiento de la ascitis suele concentrarse en la restricción de la dieta de sodio y la administración de diuréticos. También pudiera resultar necesario restringir el consumo de agua.
Debido a las muchas limitaciones en torno a la restricción del sodio, el consumo de diuréticos se ha convertido en la base del tratamiento. Se utilizan 2 clases de diuréticos: uno que actúa sobre la porción distal de la nefrona para inhibir la reabsorción de sodio dependiente de aldosterona, y un diurético de asa, como la furosemida. Con frecuencia se administran complementos de potasio por vía oral para prevenir la hipopotasemia. La postura erecta se relaciona con la activación del sistema renina-angiotensina-aldosterona; por ende, el reposo en cama podría recomendarse en personas con grandes cantidades de ascitis. La paracentesis de volumen alto (extracción de 5 L o más de líquido ascítico) puede realizarse en quienes presentan ascitis masiva y compromiso pulmonar. Dado que la eliminación del líquido genera una disminución del volumen intravascular, junto con un aumento en la actividad de la renina en el plasma y la reabsorción de sodio y agua mediada por aldosterona en los riñones, suele administrarse un expansor de volumen como la albúmina, para mantener el volumen circulante efectivo. En pacientes con ascitis resistente es posible colocar una derivación transyugular intrahepática portosistémica (DTIP).
La peritonitis bacteriana espontánea es una complicación que se observa en personas con cirrosis y ascitis. La infección es grave y conlleva una tasa de mortalidad elevada, incluso si recibe tratamiento con antibióticos. Se presume que el líquido peritoneal sufre la siembra de bacterias a partir de la sangre o la linfa, o por el paso de éstas a través de la pared intestinal.
Su sintomatología incluye fiebre, alteración del estado mental y dolor abdominal. Otros síntomas que pueden presentarse son agravamiento de la encefalopatía hepática, diarrea, hipotermia y shock. Se diagnostica con un conteo de neutrófilos de 250 células/mm 3 o más en el líquido ascítico.
Esplenomegalia
En la hipertensión portal, el bazo aumenta de tamaño en forma progresiva por efecto de la derivación de la sangre hacia la vena esplénica. El bazo distendido a menudo da origen al secuestro de un número significativo de elementos de la sangre, con el desarrollo de un síndrome conocido como hiperesplenismo, el que se caracteriza por una disminución del período de vida de todos los elementos formes de la sangre y una reducción subsecuente de sus cifras, lo que determina anemia, trombocitopenia y leucopenia. Se cree que esta disminución se debe al aumento de la velocidad de eliminación que resulta del tiempo de tránsito prolongado a través del bazo crecido.
Cortocircuitos portosistémicos
Con la obstrucción gradual del flujo venoso o en el hígado, la presión en la vena porta aumenta y se desarrollan grandes vías colaterales entre las venas porta y las sistémicas que irrigan las regiones inferiores del recto y el esófago, así como las venas umbilicales del ligamento falciforme que se inserta en la pared anterior del abdomen. Las colaterales que existen entre las venas ilíacas inferior e interna pueden dar origen a hemorroides. En algunas personas, la vena umbilical fetal no se encuentra del todo obliterada; forma un canal en la pared abdominal anterior. Las venas dilatadas en torno al ombligo se denominan cabeza de medusa. También es posible el desarrollo de cortocircuitos portopulmonares, que permiten que la sangre evite pasar por los capilares pulmonares, lo que interfiere con la oxigenación hemática y provoca a cianosis.
Desde la perspectiva clínica, los canales colaterales más importantes son los que conectan a las venas porta y las coronarias estomáquicas, que conducen a la reversión del flujo y a la formación de várices de pared delgada en la submucosa del esófago (figura 46-15). Estas várices esofágicas de pared delgada se encuentran en riesgo de ruptura, fenómeno que genera hemorragia masiva, en ocasiones mortal.
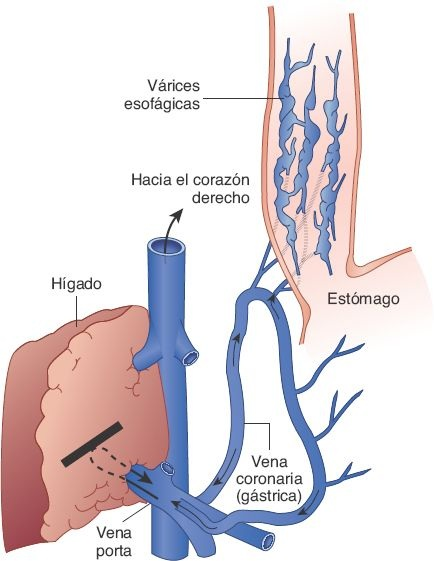
El compromiso para la síntesis hepática de factores de coagulación y la disminución en las concentraciones plaquetarias (es decir, trombocitopenia) que son consecuencia de la esplenomegalia pueden complicar aún más el control de la hemorragia esofágica. Las várices esofágicas se desarrollan en el 5% al 15% de las personas con cirrosis, y alrededor del 33% de éstas presenta hemorragia varicosa.
El tratamiento de la hipertensión portal y las várices esofágicas se dirige a la prevención de la hemorragia inicial, el control de la hemorragia aguda y la prevención de la hemorragia recurrente.
El tratamiento farmacológico se utiliza para reducir la presión venosa portal y prevenir la hemorragia inicial. Con este propósito, es común el consumo de fármacos bloqueadores β-adrenérgicos (ej. propranolol). Estos medicamentos reducen la presión venosa portal al disminuir el flujo sanguíneo esplácnico y con ello limitar el flujo de sangre a través de los canales colaterales.Existen varios métodos para controlar la hemorragia aguda, entre otros, la administración de octreótido o vasopresina, el taponamiento con balón, la escleroterapia endoscópica, la ligadura vascular y la transección esofágica. El octreótido, un análogo sintético de acción prolongada de la somatostatina, reduce el flujo sanguíneo esplácnico y hepático, así como las presiones portales en pacientes con cirrosis. La vasopresina, una hormona del lóbulo posterior de la hipófisis, es un vasoconstrictor no selectivo, que tiene potencial de generar efectos colaterales indeseables, por lo que su consumo es limitado.
Toda vez que el octreótido produce menos efectos colaterales y parece ser más efectivo que la vasopresina, se ha convertido en el medicamento de elección para el manejo farmacológico de la hemorragia varicosa aguda. El taponamiento con balón permite la compresión de las várices y se logra por medio de la inserción de una sonda con un balón inflable gástrico y otro esofágico. Una vez que la sonda se inserta, se inflan los balones; el balón esofágico comprime las várices esofágicas sangrantes, en tanto que el balón gástrico ayuda a mantener la sonda en posición. Durante la escleroterapia endoscópica se aplica en las várices una inyección con una solución esclerosante que oblitera el lumen vascular.
La prevención de la hemorragia recurrente se concentra en la disminución de la presión venosa portal y en la desviación del flujo sanguíneo, para alejarlo de los canales colaterales sensibles de ruptura. Pueden utilizarse 2 procedimientos con este propósito: la construcción quirúrgica de uncortocircuito portosistémico o la colocación de una DTIP. Los procedimientos para formación de cortocircuitos portosistémicos quirúrgicos implican la creación de una abertura entre la vena porta y una vena sistémica. Estos cortocircuitos conllevan tasas de complicaciones considerables, y la DTIP se ha convertido en el tratamiento preferido para el manejo de la hipertensión portal resistente. El procedimiento de la DTIP implica la inserción de una endoprótesis metálica expansible entre una ramificación de la vena hepática y la vena por ta, mediante el empleo de un catéter que se inserta a través de la vena yugular interna. Una limitación del procedimiento es que en la ma yoría de los casos, al pasar el tiempo se desarrollan estenosis y trombosis de la endoprótesis, lo que implica el riesgo de recurrencia del sangrado. Una complicación que se vincula con la formación de un cortocircuito portosistémico es la encefalopatía hepática que, se cree, se desarrolla cuando el amoníaco y otras sustancias nefrotóxicas derivadas de intestino pasan directamente hacia la circulación sistémica sin pasar por el hígado.
Insuficiencia hepática
La complicación clínica más grave de la hepatopatía es la insuficiencia hepática. Puede ser resultado de una destrucción súbita y masiva del hígado, como en el caso de la hepatitis fulminante, o derivar de un daño progresivo al órgano, como en la cirrosis alcohólica. Independientemente de la causa, el 80% al 90% de la capacidad funcional del hígado debe perderse para que se presente insuficiencia hepática. En muchos casos, los efectos de descompensación progresiva de la enfermedad se aceleran por condiciones intercurrentes, como hemorragia gastrointestinal, infección sistémica, trastornos electrolíticos o afecciones sobrepuestas como la insuficiencia cardíaca.
Manifestaciones clínicas
Las manifestaciones de la insuficiencia hepática corresponden a las distintas funciones de síntesis, almacenamiento, metabolismo y eliminación del hígado (figura 46-16).
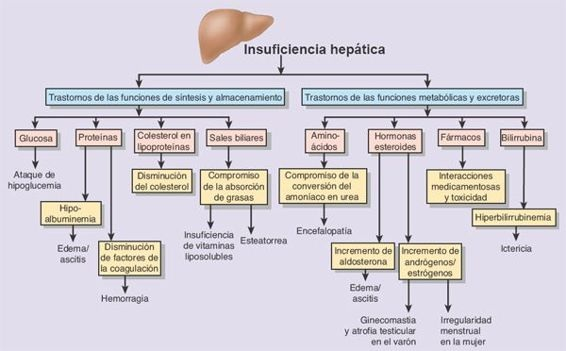
El fetor hepaticus hace referencia a un olor a humedad y dulzón del aliento de la persona con insuficiencia hepática avanzada; es consecuencia de los productos colaterales del metabolismo de las bacterias intestinales.
Trastornos hematológicos
La insuficiencia hepática puede inducir anemia, trombocitopenia, defectos de la coagulación y leucopenia. La anemia pudiera deberse a la pérdida hemática, a una destrucción excesiva de eritrocitos o al compromiso de la formación de esas células. Una insuficiencia de ácido fólico puede determinar la presencia de anemia megaloblástica grave. Los cambios en la composición lipídica de la membrana de los eritrocitos favorecen la hemólisis. Puesto que los factores V, VII, IX y X, la protrombina y el fibrinógeno se sintetizan en el hígado, su disminución en la hepatopatía incide en los trastornos hemorragíparos. La malabsorción de la vitamina K liposoluble contribuye en mayor medida al compromiso de la síntesis de estos factores de la coagulación. La trombocitopenia se presenta a menudo como consecuencia de la esplenomegalia. La persona con insuficiencia hepática se encuentra en riesgo de desarrollar púrpura, tiende a la formación de equímosis, a la hematuria y la hemorragia menstrual anómala, además de mostrar vulnerabilidad a la hemorragia a partir del esófago y de otros segmentos del tubo gastrointestinal.
Trastornos endocrinos
El hígado metaboliza las hormonas esteroides. Los trastornos endocrinos, en particular las alteraciones de la función gonadal (hormonas sexuales), son acompañantes comunes de la cirrosis y la insuficiencia hepática. Las mujeres pueden presentar irregularidades menstruales (por lo general, amenorrea), pérdida de la libido y esterilidad. En los varones, las concentraciones de testosterona suelen caer; los testículos sufren atrofia, y se presentan pérdida de la libido, impotencia y ginecomastia. Una disminución del metabolismo de la aldosterona pudiera contribuir a la retención de sal y agua en el riñón, junto con una reducción de la concentración sérica de potasio, que derivadel aumento en la eliminación de este elemento.
Trastornos cutáneos
La insuficiencia hepática trae consigo múltiples trastornos cutáneos. Estas lesiones, llamadas de manera indistinta arañas vasculares, telangiectasias, angiomas en araña y nevos en araña, se observan la mayoría de las veces en el hemicuerpo superior. Están conformadas por una arteriola pulsátil central a partir de la que irradian vasos más pequeños. El eritema palmar consiste en el enrojecimiento de las palmas, quizá por un aumento del flujo sanguíneo secundario al incremento del gasto cardíaco. Puede observarse el desarrollo de dedos en palillo de tambor en personas con cirrosis. La ictericia suele ser una manifestación tardía de la insuficiencia hepática.
Síndrome hepatorrenal
El síndrome hepatorrenal alude a una insuficiencia renal funcional, que en ocasiones se observa durante las fases terminales de la insuficiencia hepática sin que existan causas funcionales de enfermedad renal. Se caracteriza por azoemia progresiva, incremento de creatinina en las concentraciones séricas y oliguria. Si bien su causa básica se desconoce, se cree que una disminución del flujo sanguíneo renal desempeña algún papel. Por último, cuando la insuficiencia renal se sobrepone a la hepática, se presentan azoemia y elevación en las concentraciones del amoníaco en la sangre; se piensa que esta condición contribuye a la encefalopatía hepática y al coma.
Encefalopatía hepática
La encefalopatía hepática se refiere a todas las manifestaciones del sistema nervioso central en la insuficiencia hepática. Se caracteriza por trastornos neurológicos, que van desde la falta de alerta hasta la confusión, el coma y las convulsiones. Un signo muy temprano de la encefalopatía hepática es un temblor aleteante, denominado asterixis. Pueden presentarse distintos grados de pérdida de la memoria, aunados a cambios en la personalidad, como euforia, irritabilidad, ansiedad y falta de preocupación sobre el aspecto personal y la persona en general. El lenguaje puede comprometerse, y el paciente pudiera ser incapaz de llevar a cabo ciertos movimientos voluntarios.
La encefalopatía puede evolucionar hasta la rigidez de descerebración y luego a un coma profundo terminal.
Si bien se desconoce la causa de la enfermedad, se cree que un factor que incide en ésta es la acumulación de neurotoxinas, que aparecen en la sangre debido a que el hígado pierde su capacidad de destoxificación. Una de las neurotoxinas sospechosas es el amoníaco. Una función en particularimportante del hígado es la conversión del amoníaco, un producto colateral del metabolismo de las proteínas y los aminoácidos, en urea. El ion amonio se produce en abundancia en el tubo digestivo, en especial en el colon, por la degradación bacteriana de las proteínas y los aminoácidos contenidos en el lumen. Por lo regular, estos iones amonio se difunden hacia la sangre portal y se transportan hacia el hígado, donde se convierten en urea antes de ingresar a la circulación general. Cuando la sangre que proviene del intestino puentea al hígado, o cuando el hígado es incapaz de convertir el amoníaco en urea, éste llega en forma directa a la circulación general y de ahí alcanza la circulación cerebral. La encefalopatía hepática pudiera agravarse después de una comida rica en proteínas o una hemorragia del tubo digestivo. Los narcóticos y los tranquilizantes se metabolizan de manera deficiente en el hígado, y la administración de estos medicamentos puede inducir depresión del sistema nervioso central y precipitar la encefalopatía hepática.
Un antibiótico no absorbible como la neomicina puede administrarse para erradicar las bacterias del intestino y así prevenir la síntesis de amoníaco. Otro fármaco que puede suministrarse es la lactulosa. No se absorbe en el intestino delgado sino que se desplaza en forma directa hasta el colon, donde se cataboliza por la acción de las bacterias colónicas para obtener ácidos orgánicos pequeños que generan la producción de heces abundantes, acuosas, con un pH bajo. El pH bajo favorece la conversión del amoníaco en iones amonio, que no ingresan a la sangre. El pH ácido también inhibe la degradación intestinal de los aminoácidos, las proteínas y la sangre.
Tratamiento
El tratamiento de la insuficiencia hepática se dirige a eliminar el consumo de alcohol cuando la condición se debe a cirrosis alcohólica, a la prevención de infecciones, a la provisión de carbohidratos y calorías suficientes para prevenir la degradación de las proteínas, a la corrección de los desequilibrios hidroelectrolíticos y en particular la hipopotasemia, y a la disminución de la producción de amoníaco en el tubo digestivo mediante el control de consumo de proteínas.
En muchos casos, el trasplante hepático sigue siendo el único tratamiento efectivo. El trasplante hepático se está convirtiendo con rapidez en una forma realista de tratamiento para muchas personas con hepatopatía crónica irreversible, insuficiencia hepática fulminante, cirrosis biliar primaria, hepatitis crónica activa, colangitis esclerosante y ciertos trastornos metabólicos que generan hepatopatía en fase terminal. En 2009 la tasa de sobrevivencia a 5 años en Estados Unidos era del 74% y el 79% para personas que recibían hígados de donadores cadavéricos y vivos, respectivamente.
Además de una sobrevivencia mayor, los receptores de trasplante hepático experimentan ahora una mejoría en la calidad de vida, lo que incluye su regreso al empleo activo. Desafortunadamente, la escasez de donadores limita el número de trasplantes que se realizan y muchas personas mueren cada año mientras esperan un trasplante. En la actualidad existen 16.000 personas en lista de espera para la recepción de un trasplante hepático en Estados Unidos. Durante los últimos años se desarrollaron distintos métodos innovadores para resolver la escasez, lo que incluye el trasplante hepático dividido, en que un hígado cadavérico se separa en 2 partes que se trasplantan a los receptores, y el trasplante de donadores vivos, en el que un segmento o lóbulo del hígado de un donador vivo se reseca e injerta en un receptor.
Cáncer hepático
Cánceres primarios del hígado
Existen 2 tipos principales de cáncer hepático primario: el carcinoma hepatocelular, que se origina a partir de los hepatocitos, y el colangiocarcinoma, un cáncer de las células de los conductos biliares.
Carcinoma hepatocelular
En 2010, en Estados Unidos el cáncer hepatocelular generó 19.000 de los casos nuevos de cáncer hepático, lo que lo convirtió en la variante más frecuente de cáncer hepático. En regiones donde existen mayores recursos, como Europa, Australia y Estados Unidos, la incidencia es de entre 2,5 y 5 casos por 100.000. En Tailandia, Corea, Japón y China se han identificado tasas hasta de 40 casos por 10.000. También se ha encontrado aumento en la incidencia en países desarrollados, como consecuencia de la infección crónica por VHC. En Estados Unidos la frecuencia se incrementó de 1,4 casos por 100.000 entre 1976 y 1980, hasta 2,4 casos entre 1990 y 1995. Si bien los tumores primarios del hígado son relativamente raros en los países desarrollados, el hígado comparte con el pulmón la característica de ser la ubicación más frecuente de los tumores metastásicos.
Entre los factores que se han identificado como etiología del cáncer hepático se encuentran la hepatitis viral crónica (es decir, por VHB, VHC y VHD), la cirrosis, la exposición a largo plazo a factores ambientales como la aflatoxina, y el consumo de agua contaminada con arsénico. La forma en que estos agentes etiológicos contribuyen al desarrollo del cáncer hepático aún no se esclarece. En el caso del VHB y el VHC, que se integran al ADN del hospedero, los ciclos repetidos de muerte celular y regeneración determinan un potencial para el desarrollo de mutaciones productoras de cáncer.
En ciertas áreas donde el carcinoma hepatocelular es endémico, las aflatoxinas (sintetizadas por mohos de los desperdicios de comida), como Aspergillus flavus y Aspergillus parasiticus, son agentes carcinógenos con particular potencia. Se activan en los hepatocitos y sus productos se incorporan al ADN del hospedero, lo que determina un potencial para el desarrollo de mutaciones inductoras de cáncer. Un sitio en especial susceptible para la mutación inducida por aflatoxinas es el gen supresor tumoral TP53.
Manifestaciones clínicas y diagnóstico
Las manifestaciones del cáncer hepatocelular son con frecuencia insidiosas en su inicio y se encuentran enmascaradas por las derivadas de cirrosis o hepatitis crónica. Los síntomas iniciales incluyen debilidad, anorexia, pérdida ponderal, fatiga, aumento del volumen abdominal, sensación de plenitud abdominal, y un dolor abdominal sordo y persistente. La ascitis, que muchas veces impide la detección de la pérdida ponderal, también es común. La ictericia, en caso de existir, suele ser leve. Puede observarse un incremento rápido del tamaño del hígado y la intensificación de la ascitis en personas con cirrosis preexistente. Suele haber hepatomegalia cuando estos síntomas aparecen.
Distintos síndromes paraneoplásicos (es decir, trastornos derivados de la síntesis ectópica de hormonas o de factores de crecimiento en el tumor) se han relacionado con el cáncer hepatocelular, e incluyen el desarrollo de eritrocitosis (eritropoyetina), hipoglucemia (factor de crecimiento similar a la insulina) e hipercalcemia (proteína relacionada con la paratiroides).
La α-fetoproteína puede identificarse a lo largo de la vida fetal, pero rara vez es detectable en el suero después de los 2 años de edad. Cuando se identifican en el adulto concentraciones altas de α-fetoproteína, suelen ser indicativas de la existencia del carcinoma hepatocelular; no obstante, no todos los cánceres hepáticos primarios la sintetizan. De acuerdo con lo anterior, se recomienda la utilización de métodos adicionales para el diagnóstico, como ecografía, TC y IRM. Puede recurrirse a la biopsia hepática para confirmar el diagnóstico.
Tratamiento
Los cánceres primarios del hígado suelen encontrarse muy avanzados en el momento del diagnóstico. El tratamiento de elección consiste en la hepatectomía subtotal, en caso de que la condición lo permita. La quimioterapia y la radioterapia son en gran medida paliativas. Si bien el trasplante hepático puede constituir una opción en personas con cirrosis bien compensada y tumores pequeños, a menudo resulta impráctica debido a la escasez de órganos donados.
Colangiocarcinoma
El colangiocarcinoma, con una incidencia de 1,2 a 0,5 por 100.000 en Norteamérica, se presenta con mucha menor frecuencia que el carcinoma hepatocelular. La etiología, las características clínicas y el pronóstico varían en grado considerable en relación con la región del árbol biliar de la que se origina el tumor. El colangiocarcinoma se asocia con factores de riesgo distintos a los del carcinoma hepatocelular, y casi todos se vinculan con la inflamación crónica y la lesión del epitelio de los conductos biliares. El colangiocarcinoma muchas veces se manifiesta por dolor, pérdida ponderal, anorexia, aumento del volumen abdominal o sensación de masa en el hipocondrio derecho. Los tumores que afectan los conductos biliares centrales o distales pueden presentarse con ictericia.
Tumores metastásicos
Los tumores metastásicos del hígado son mucho más comunes que los tumores primarios. Entre los cánceres primarios que los originan, con frecuencia se encuentran el cáncer colorrectal y los que se diseminan a partir de la mama, colon, pulmón y el sistema urogenital. Además, algunos tumores de origen neuroendocrino se diseminan hacia el hígado. Con frecuencia es difícil distinguir los tumores primarios de los metastásicos mediante el empleo de TC, IRM o ecografía. El diagnóstico se confirma casi siempre con biopsia.
Trastornos de la vesícula biliar y el páncreas exocrino
Trastornos de la vesícula biliar y los conductos biliares extrahepáticos
El sistema hepatobiliar está conformado por la vesícula biliar; los conductos hepáticos izquierdo y derecho, que se unen para constituir el conducto hepático común; el conducto cístico, que se extiende hacia la vesícula biliar, y el conducto biliar común, o colédoco, compuesto por la unión del conducto hepático común y el conducto cístico (figura 46-17).
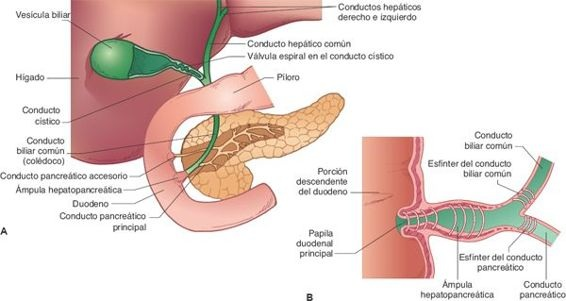
El conducto biliar común desciende por detrás de la primera porción del duodeno, donde entra en contacto con el conducto pancreático principal. Estos conductos se unen para conformar el ámpula hepatopancreática. El músculo circular que se ubica en torno al extremo distal del conducto biliar común se engrosa para constituir un esfínter para esa estructura.
La vesícula biliar es un saco muscular distensible y con forma de pera que se aloja en la cara inferior del hígado. Tiene una capa serosa peritoneal externa, una capa intermedia de músculo liso y una capa mucosa interna que tiene continuidad con el recubrimiento del conducto biliar. La función de la vesícula es almacenar y concentrar la bilis. La bilis contiene sales biliares, colesterol, bilirrubina, lecitina, ácidos grasos, agua y los electrolitos que por lo regular se encuentran en el plasma. El colesterol de la bilis carece de función conocida. Se asume que es un producto colateral de la formación de sales biliares, y su presencia se vincula con la función de excreción de la bilis. Por común insoluble en agua, el colesterol se vuelve soluble por la acción de las sales biliares y la lecitina, que se combinan con él para formar micelios. En la vesícula biliar, el agua y los electrolitos se absorben a partir de la bilis hepática, lo que hace que la bilis se concentre. Dado que ni la lecitina ni las sales biliares se absorben en la vesícula biliar, se concentran a la par del colesterol; de esta manera, se conserva la solubilidad del colesterol.
El ingreso del alimento al intestino hace que la vesícula biliar se contraiga y que el esfínter del conducto biliar común se relaje, de tal manera que la bilis almacenada en la vesícula se desplaza hacia el duodeno. El estímulo para la contracción de la vesícula biliar es sobre todo hormonal. Los productos de la digestión de los alimentos, en especial los lípidos, estimulan la liberación de la hormona gastrointestinal colecistocinina a partir de la mucosa del duodeno, que provee un estímulo intenso para la contracción de la vesícula biliar. El papel de otras hormonas gastrointestinales en la liberación de la bilis se conoce con menos claridad.
La presión en el conducto biliar común es responsable en gran medida de regular el paso de la bilis hacia el intestino. En condiciones normales, la vesícula biliar regula esta presión. Colecta y almacena la bilis al tiempo que se relaja y la presión en el conducto biliar común disminuye, y luego vacía la bilis al intestino cuando se contrae, lo que genera un aumento de la presión en el conducto biliar común. Tras la extirpación quirúrgica de la vesícula biliar, la presión en el conducto biliar común se modifica y hace que la estructura se dilate. Los esfínteres del conducto biliar común regulan entonces el flujo biliar.
Dos trastornos frecuentes del sistema biliar son la colelitiasis (es decir, formación de litos vesiculares) y la inflamación de la vesícula biliar (colecistitis) o del conducto biliar común (colangitis). En las poblaciones occidentales adultas, el 15% de las personas presenta litiasis vesicular. En ambas circunstancias, la hipersecreción del colesterol biliar parece desempeñar un papel importante.
Colelitiasis
La colelitiasis o litiasis vesicular se debe a la precipitación de sustancias que contiene la bilis, de manera primordial colesterol y bilirrubina. Alrededor del 80% de los litos vesiculares se encuentra conformado por colesterol; el otro 20% lo integran litos de pigmento negro o pardo, compuestos por glucoproteínas de mucina y sales de calcio. Muchos litos tienen composición mixta. En la figura 46-18 se muestra una vesícula biliar con numerosos litos de colesterol.

Dos factores principales contribuyen a la formación de los litos vesiculares: las anomalías de la composición de la bilis (en particular, aumento del colesterol) y la estasia biliar. La formación de los litos de colesterol se asocia con la obesidad, y se presenta con más frecuencia en mujeres, en particular en aquéllas que han tenido embarazos múltiples o están tomando anticonceptivos orales.
Todos estos factores hacen que el hígado excrete más colesterol a la bilis. Los estrógenos reducen la síntesis de ácidos biliares en las mujeres. Se cree que el lodo biliar (mucoproteína espesa en la vesícula biliar con cristales diminutos de colesterol atrapados) es el precursor de los litos vesiculares. El lodo se observa a menudo durante el embarazo, los estados de inanición y la pérdida ponderal rápida. Los medicamentos que reducen las concentraciones de colesterol en el suero, como el clofibrato, también generan un aumento en la excreción del colesterol a la bilis.
Los trastornos por malabsorción que derivan de la enfermedad ileal o la cirugía de puenteo intestinal, por ejemplo, tienden a interferir con la absorción de las sales biliares, necesarias para mantener la solubilidad del colesterol. La inflamación de la vesícula biliar altera las características de absorción de la capa mucosa, lo que propicia que se presente una absorción excesiva de agua y sales biliares. Los litos vesiculares de colesterol son en extremo frecuentes entre los indios americanos, los indios chilenos y los chilenos, lo que sugiere que algún componente genético pudiera desempeñar un papel en laformación de los litos vesiculares. Los litos pigmentarios que contienen bilirrubina se identifican en personas con enfermedad hemolítica (ej. enfermedad de células falciformes) y cirrosis hepática.
Manifestaciones clínicas
Muchas personas con litos vesiculares carecen de síntomas. Los litos vesiculares inducen síntomas cuando obstruyen el flujo biliar o inducen inflamación. Los litos pequeños (es decir, <8 mm de diámetro) pasan hacia el conducto biliar común, y generan síntomas dispépticos y cólico biliar. Los litos más grandes tienen más probabilidad de obstruir el flujo e inducir ictericia. El dolor del cólico biliar suele ubicarse en el cuadrante superior derecho o en el área epigástrica, y puede referirse en la región superior de la espalda, el hombro derecho o la región interescapular. Por lo general, el dolor es de inicio abrupto, se incrementa de manera constante en intensidad, persiste entre 30 min y 5 h, y va seguido por una sensación de dolor en el cuadrante superior derecho.
Colecistitis aguda y crónica
La colecistitis aguda es una inflamación aguda de la vesícu la biliar, por lo regular secundaria a la obstrucción de la vía de salida de la vesícula. La mayoría de los casos de colecistitis aguda (del 85% al 90%) se relaciona con la presencia de litos vesiculares (colecistitis litiásica). El resto (colecistitis acalculosa), con septicemia, traumatismo grave o infección de la vesícula biliar. Existe la teoría de que la obstrucción del conducto cístico por un lito vesicular origina la liberación de fosfolipasa a partir del epitelio de la vesícula biliar. A su vez, esta enzima puede hidrolizar la lecitina y liberar lisolecitina, una toxina con actividad contra la membrana. Al mismo tiempo, la disrupción del recubrimiento mucoso protector normal del epitelio deja a las células de la mucosa vulnerables al daño, debido a la acción detergente de las sales biliares concentradas. La colecistitis acalculosa aguda se desarrolla sin causa aparente en el 50% de los casos; el traumatismo, las quemaduras, el lodo biliar y la vasculitis son algunos de los posibles factores etiológicos. La colecistitis acalculosa aguda puede evolucionar con rapidez a gangrena y perforación, puesto que el proceso parece implicar un infarto transmural, más que cambios inflamatorios relacionados con la litiasis.
La colecistitis crónica se origina a partir de episodios repetidos de colecistitis aguda o irritación crónica de la vesícula biliar por la presencia de litos. Se caracteriza por grados variables de inflamación crónica. Casi siempre existen litos vesiculares. La colelitiasis con colecistitis crónica puede relacionarse con exacerbaciones agudas de inflamación de la vesícula biliar, coledocolitiasis, pancreatitis y, rara vez, carcinoma de la vesícula biliar.
Manifestaciones clínicas
Las personas con colecistitis aguda suelen experimentar dolor de inicio agudo en el cuadrante superior derecho o el epigastrio, muchas veces acompañado de febrícula, anorexia, náuseas y vómito. Mientras que el cólico biliar por obstrucción del conducto cístico es transitorio, en la colecistitis aguda el dolor es persistente. Las personas con colecistitis litiásica suelen haber experimentado episodios previos de dolor biliar, aunque esto no siempre sucede. El dolor puede aparecer en forma sorpresiva, y determinar una urgencia quirúrgica. En ausencia de atención médica, la crisis suele ceder en un período que va de 7 a 10 días. En personas que se recuperan, la recurrencia es frecuente.
El inicio de la colecistitis acalculosa tiende a ser más insidioso debido a que a sus manifestaciones se sobreponen a las condiciones subyacentes precipitantes de la crisis. En personas con enfermedad grave resulta crucial el reconocimiento temprano, puesto que la tardanza para el tratamiento puede poner en riesgo la vida. Las personas con colecistitis aguda a menudo presentan elevación en el conteo leucocitario, y muestran aumentos leves de ATS, ATL, fosfatasa alcalina y bilirrubina.Las manifestaciones de la colecistitis crónica son más vagas que las de la colecistitis aguda.
Puede existir intolerancia a los alimentos grasosos, aerofagia y otras indicaciones de malestar. Al presentarse obstrucción al flujo biliar, derivada de los litos vesicales, con frecuencia se observan episodios de dolor crónico. La vesícula biliar, que en la colecistitis crónica suele contener litos, puede experimentar aumento o disminución de volumen, o tener tamaño normal.
Diagnóstico y tratamiento
Las técnicas que se utilizan para el diagnóstico de la enfermedad de la vesícula biliar incluyen ecografía, centelleografía vesicular (gammagrafía nuclear) y estudios de TC. La ecografía se usa en forma amplia para este propósito y ha sustituido en gran medida a la colecistografía oral en la mayoría de los centros médicos. Puede detectar litos de hasta 1 cm a 2 cm, y su precisión general para la detección de la enfermedad vesicular es alta. Además de los litos, la ecografía puede detectar el engrosamiento de la pared, que revela inflamación. También permite descartar otras causas de dolor en el cuadrante superior derecho, como los tumores.
La centelleografía vesicular, también denominada gammagrafía vesicular, depende de la capacidad del hígado para extraer un radionúclido que se inyecta con rapidez, el tecnesio 99m, unido a un ácido iminodiacético, que se excreta hacia los conductos biliares. Se obtienen imágenes seriadas en pocos minutos tras la inyección del marcador, y cada 10 min a 15 min durante la siguiente hora. La gammagrafía vesicular es muy preciso para la detección de la colecistitis aguda. Si bien la TC no es tan precisa como la ecografía para detectar los litos biliares, puede revelar el engrosamiento de la pared vesicular o la presencia de líquido pericolecístico relacionado con la colecistitis aguda.
La enfermedad de la vesícula biliar suele tratarse mediante la extirpación del órgano. La vesícula biliar almacena y concentra la bilis y, por lo regular, su extirpación no interfiere con la digestión. La colecistectomía laparoscópica se ha convertido en el tratamiento de elección para la enfermedad vesicular sintomática. El procedimiento implica la inserción de un laparoscopio a través de una incisión pequeña cerca del ombligo, mientras que los instrumentos quirúrgicos se introducen por varias heridas realizadas con bisturí en el hemiabdomen superior. Aunque esta opción implica más tiempo que el antiguo procedimiento quirúrgico abierto, suele necesitar sólo una noche de hospitalización. Una ventaja importante del colecistectomía laparoscópica es que las personas pueden reincorporarse al trabajo en una o 2 semanas, en comparación con las 4 a 6 semanas que se requieren tras la colecistectomía abierta.
Coledocolitiasis y colangitis
La coledocolitiasis alude a la presencia de litos en el conducto biliar común (colédoco), en tanto que la colangitis se refiere a la inflamación de la misma estructura. En general, los litos en el conducto biliar común se originan en la vesícula biliar, pero pueden formarse de manera espontánea en el propio conducto.
Manifestaciones clínicas
Las manifestaciones de la coledocolitiasis son similares a las de la litiasis vesicular y la colecistitis aguda. Existe antecedente de cólico biliar agudo y dolor en el cuadrante superior derecho del abdomen, con escalofríos, fiebre e ictericia relacionados con los cuadros de dolor abdominal. En caso de obstrucción del conducto biliar común se detectan bilirrubinuria e hiperbilirrubinemia. Entre las complicaciones se encuentra la colangitis supurativa aguda, que se acompaña de líquido purulento en el conducto biliar común. Se caracteriza por el desarrollo de anomalías del sensorio, letargo y shock séptico. La colangitis supurativa aguda representa una urgencia endoscópica o quirúrgica. Los litos en el conducto biliar común también pueden obstruir el flujo de salida del conducto pancreático y desencadenar pancreatitis secundaria.
Diagnóstico y tratamiento
La ecografía, la TC y los estudios de imagen con radionúclidos pueden utilizarse para revelar la dilatación de los conductos biliares y el compromiso del flujo sanguíneo. La ecografía endoscópica y la colangiografía por resonancia magnética se usan para detectar los litos en el conducto biliar común. Tanto la colangiografía transhepática percutánea (CTP) como la colangiopacreatografía retrógrada endoscópica (CPRE) constituyen un medio directo para determinar la causa, la ubicación y el grado de la obstrucción. La CTP implica la inyección de una tinción en forma directa dentro del árbol biliar. Para esto se requiere la inserción de una aguja delgada y flexible a través de una incisión pequeña realizada en la piel, que se avanza hasta el interior del árbol biliar. La CPRE implica el paso de un endoscopio hasta el duodeno y la inserción de una sonda a través del ámpula hepatopancreática. La CPRE puede emplearse para aumentar el diámetro de la abertura del esfínter del ámpula hepatopancreática, lo que pudiera permitir el paso del lito atrapado, o la inserción de un instrumento en el conducto biliar común para extraer el lito.
En personas con colelitiasis los litos del colédoco suelen manejarse mediante extracción, a la que sigue una colecistectomía laparoscópica. Para el control de la infección se administra tratamiento antibiótico, con un fármaco que se excrete en la bilis. La descompresión de urgencia del conducto biliar común, por lo general mediante CPRE, pudiera ser necesaria en pacientes sépticos o que no mejoran con el tratamiento antibiótico.
Cáncer de la vesícula biliar
El cáncer de la vesícula biliar es el quinto en frecuencia entre los del tubo gastrointestinal. Es un poco más común en mujeres y se presenta con más frecuencia durante la séptima década de la vida. El inicio de los síntomas suele ser insidioso, y se asemejan a los de la colecistitis. Muchas veces, el diagnóstico se establece de manera inesperada en el momento de una cirugía de vesícula biliar.
Alrededor del 70% al 80% de las personas con cáncer de la vesícula biliar presenta colelitiasis.
Dada su capacidad para inducir irritación crónica en la mucosa de la vesícula, se piensa que la colelitiasis participa en el desarrollo del cáncer vesicular. La tasa de sobrevivencia a 5 años varía según el estadio del carcinoma, con una sobrevivencia del 80% en el estadio 0 y del 2% en el estadio IVB.
Trastornos del páncreas exocrino
El páncreas se encuentra dispuesto en sentido transversal, en la parte posterior del hemiabdomen superior (figura 46-1). La cabeza del páncreas se ubica a la derecha del abdomen; descansa contra la curva del duodeno, en la región del ámpula hepatopancreática y su entrada al duodeno. El cuerpo del páncreas se localiza por debajo del estómago. La cola está en contacto con el bazo. El páncreas se encuentra prácticamente escondido debido a su ubicación posterior; a diferencia de muchos otros órganos, no puede palparse. Debido a su posición y a su gran reserva funcional, los síntomas de afecciones como el cáncer del páncreas no suelen aparecer hasta que el trastorno está muy avanzado.
El páncreas es un órgano tanto endocrino como exocrino. El páncreas exocrino está formado por lobulillos constituidos por células acinares, que secretan enzimas digestivas hacia un sistema de conductos microscópicos. Estos conductos drenan en el conducto pancreático principal, que se extiende de izquierda a derecha a todo lo largo de este órgano. El conducto pancreático principal y el conducto biliar común se unen para constituir el ámpula hepatopancreática, que drena en el duodeno.
El esfínter del conducto pancreático controla el flujo de las secreciones pancreáticas hacia el duodeno (figura 46-17).
Las secreciones pancreáticas contienen enzimas proteolíticas que degradan las proteínas de ladieta, entre otras, tripsina, quimiotripsina, carboxipeptidasa, ribonucleasa y desoxirribonucleasa. El páncreas también secreta amilasa pancreática, que degrada los almidones, y lipasas, que hidrolizan las grasas neutras en glicerol y ácidos grasos. Las enzimas pancreáticas se secretan en forma inactiva y se activan en el intestino. Esto es im portante porque las enzimas digerirían el tejido del páncreas mismo si se secretaran en forma activa. Las células acinares secretan un inhibidor de la tripsina, que impide la activación de esta enzima. Puesto que la tripsina activa otras enzimas proteolíticas, el inhibidor de la tripsina impide la activación subsecuente de las otras enzimas.
Dos tipos de enfermedad pancreática se analizan en este capítulo: la pancreatitis aguda y la crónica, y el cáncer del páncreas.
Pancreatitis aguda
Representa un proceso inflamatorio reversible de los acinos pancreáticos, que se genera por la activación prematura de las enzimas pancreáticas. Aunque el proceso de enfermedad puede limitarse al tejido pancreático, también puede implicar a los tejidos peripancreáticos o a los de órganos distantes. En Estados Unidos, hasta 220.000 personas ingresan al hospital cada año por pancreatitis aguda.
La patogénesis de la pancreatitis aguda implica la autodigestión del tejido pancreático por la acción de enzimas pancreáticas que se activan de manera inapropiada. Se sospecha que el proceso comienza con la activación de la tripsina. Una vez activada, la tripsina puede activar distintas enzimas digestivas que inducen lesión pancreática, lo que origina una respuesta inflamatoria intensa. La respuesta inflamatoria aguda misma causa daño tisular sustancial y puede extenderse más allá del páncreas, para generar un síndrome de respuesta inflamatoria sistémica y fallo multiorgánico. No obstante, varios factores se asocian al desarrollo de la pancreatitis aguda; la mayoría de los casos se debe a los litos vesiculares (litos en el conducto biliar común) o al consumo de alcohol.
En el caso de la obstrucción de la vía biliar por la presencia de litos vesiculares, se cree que la obstrucción del conducto pancreático o el reflujo biliar activan las enzimas dentro del sistema de conductos pancreáticos. Los mecanismos precisos por los que el alcohol ejerce su acción se desconocen en gran medida. La capacidad que tiene el páncreas de participar en el metabolismo oxidativo y no oxidativo del etanol y los productos colaterales lesivos que derivan de esos procesos se han relacionado con el proceso patológico. Un estudio reciente que analizó la cerveza de manera específica demostró que las secreciones pancreáticas pueden ser estimuladas por los ingredientes no alcohólicos de la bebida. La pancreatitis aguda también se relaciona con hiperlipidemia, hipercalcemia, infecciones (en particular, virales), traumatismo abdominal y quirúrgico, y medicamentos como los diuréticos tiazídicos.
Manifestaciones clínicas
Las manifestaciones de la pancreatitis aguda pueden variar desde las leves con disfunción orgánica mínima hasta las graves y que ponen en riesgo la vida. En general, alrededor del 20% de las personas con pancreatitis aguda tiene una evolución grave. El dolor abdominal es una manifestación cardinal de la enfermedad; suele ubicarse en el epigastrio o en la región periumbilical, y puede irradiarse a la espalda, el tórax o los flancos. La exploración física genera hallazgos variables, que incluyen fiebre, taquicardia, hipotensión, hipersensibilidad intensa a la palpación abdominal, dificultad respiratoria y distensión abdominal.
Entre los marcadores reconocidos de la enfermedad grave se encuentran los valores de laboratorio que cuantifican la respuesta inflamatoria (ej. proteína C reactiva), los sistemas de calificación que valoran la inflamación o el fallo orgánico y los hallazgos en los estudios de imagen. Los hallazgos clínicos como sed, gasto urinario deficiente, taquicardia progresiva, taquipnea, hipoxemia, agitación,confusión, incremento del hematócrito y ausencia de mejoría sintomática en el transcurso de 48 h, son signos de alerta de enfermedad grave inminente. Entre sus complicaciones se citan la respuesta inflamatoria sistémica, el síndrome de dificultad respiratoria aguda, la necrosis tubular aguda y el fallo orgánico. Un trastorno importante relacionado con la pancreatitis aguda es la pérdida de un gran volumen de líquido que escapa hacia los espacios retroperitoneal y peripancreático, así como a la cavidad abdominal.
Diagnóstico y tratamiento
La amilasa sérica y la lipasa son los marcadores de laboratorio que se utilizan con más frecuencia para establecer el diagnóstico de pancreatitis aguda. La lipasa sérica puede permanecer elevada un tiempo un poco mayor que la amilasa. Sin embargo, el nivel de elevación de la amilasa sérica o la lipasa no se correlaciona con la gravedad del trastorno. El conteo leucocitario puede mostrar aumento, y pueden existir hiperglucemia e hiperbilirrubinemia. La determinación de la etiología es importante para orientar el manejo inmediato y prevenir la recurrencia.
La ecografía abdominal suele realizarse para descartar la presencia de litos biliares. Los estudios de TC y la TC dinámica con contraste del páncreas se emplean para detectar la necrosis y la acumulación de líquidos. Una investigación reciente se concentró en el análisis de biomarcadores potenciales para establecer la gravedad y el pronóstico de la pancreatitis. Un estudio analizó al tripsinógeno tipo 2 y a las proteasas pancreáticas, enzimas implicadas en los procesos de autodigestión. Otros marcadores serológicos en investigación son el factor de necrosis tumoral, la proteína C reactiva, la procalcitonina, la fosfolipasa A 2 y las citocinas interleucina 8 e interleucina 10.
La estrategia terapéutica depende de la gravedad del trastorno. Las personas que presentan dolor persistente o intenso, vómito, deshidratación o signos de pancreatitis aguda grave inminente requieren hospitalización. Las medidas terapéuticas se dirigen al alivio del dolor, a «poner en reposo el páncreas» al diferir la alimentación y la hidratación orales, y a la recuperación del volumen plasmático perdido. Suele administrarse meperidina y no morfina para aliviar el dolor, puesto que genera menos espasmos del esfínter del conducto pancreático. Se establece succión gástrica para manejar la distensión intestinal y prevenir la estimulación adicional para la secreción de enzimas pancreáticas. Se administran líquidos y electrolitos intravenosos para restituir los perdidos a partir de la circulación, y combatir la hipotensión y el shock. Se infunden soluciones coloides intravenosas para restituir el líquido secuestrado en el abdomen y el espacio retroperitoneal.
Complicaciones
En personas que sobreviven a un cuadro de pancreatitis aguda grave, las complicaciones incluyen la formación de acumulaciones líquidas y la infección. En personas con pancreatitis necrosante aguda, el detrito necrótico se infecta, por lo general por microorganismos gramnegativos derivados del tubo digestivo, lo que complica la condición en mayor medida. La acumulación líquidas con una concentración alta de enzimas pancreáticas suelen relacionarse con la pérdida de continuidad del conducto pancreático y de manera eventual convertirse en seudoquistes (una acumulaciones de líquido pancreático incluida en una capa de tejido inflamatorio). La mayoría de las veces el seudoquiste se conecta con un conducto pancreático, de tal manera que su masa sigue incrementándose. Los síntomas dependen de su ubicación; por ejemplo, puede presentarse ictericia cuando un quiste se desarrolla cerca de la cabeza del páncreas, en cercanía al conducto biliar común.
Los seudoquistes pueden resolverse o, en caso de persistir, requerir intervención quirúrgica.
Pancreatitis crónica
Se caracteriza por la destrucción progresiva del páncreas exocrino, por fibrosis y, en las fases posteriores, por la destrucción del páncreas endocrino. Casi todos los factores que inducenpancreatitis aguda también tienen capacidad de inducir pancreatitis crónica. Sin embargo, existe una diferencia importante entre estas 2 condiciones, que consiste en la irreversibilidad de la disfunción pancreática característica de la pancreatitis crónica.
En los países occidentales la etiología más común de pancreatitis crónica es, por mucho, el consumo crónico de alcohol. Otras causas menos frecuentes son la obstrucción a largo plazo del conducto pancreático por seudoquistes, cálculos o neoplasias; la pancreatitis crónica autoinmunitaria, que se presenta junto con trastornos autoinmunitarios como el síndrome de Sjögren, la colangitis esclerosante primaria y la enfermedad intestinal inflamatoria; la pancreatitis crónica idiopática, que se asocia a la fibrosis quística, y la pancreatitis hereditaria, un trastorno autosómico dominante poco frecuente, vinculado con pancreatitis, tanto aguda como crónica.
Manifestaciones clínicas
La pancreatitis crónica se manifiesta por episodios similares a los de la pancreatitis aguda, aunque con menor gravedad. Las personas con pancreatitis crónica presentan episodios persistentes y recurrentes de dolor en epigastrio y en el cuadrante superior izquierdo; las crisis a menudo se precipitan por el consumo excesivo de alcohol y una alimentación desmesurada.
Anorexia, náuseas, vómito, constipación y flatulencia son comunes. De manera esporádica, la enfermedad avanza hasta el grado de generar deficiencias en las funciones pancreáticas endocrina y exocrina. En ese momento se hacen evidentes signos de diabetes mellitus y del síndrome de malabsorción (ej. pérdida ponderal y esteatorrea).
Tratamiento
El tratamiento consiste en medidas para el manejo de la patología coexistente de las vías biliares. Suele prescribirse una dieta baja en grasas. Los signos de malabsorción pueden tratarse con enzimas pancreáticas. Cuando existe diabetes, se maneja con insulina. El alcohol se prohíbe debido a que con frecuencia precipita los ataques. Puesto que existen cuadros frecuentes de dolor, la adicción a narcóticos es un problema potencial en personas con pancreatitis crónica. En ocasiones se requiere una intervención quirúrgica para aliviar el dolor y suele concentrarse en resolver cualquier obstrucción que pudiera existir. En los casos avanzados pudiera hacerse necesaria una pancreatectomía subtotal o total.
Cáncer del páncreas
El cáncer pancreático sigue siendo la cuarta causa de muerte por cáncer en Estados Unidos, con más de 44.000 casos nuevos diagnosticados cada año. Considerado como una de las afecciones malignas más mortales, el cáncer pancreático se relaciona con una tasa de sobrevivencia a 5 años de tan sólo el 4%. La incidencia de la enfermedad parece ir en aumento en todos los países estudiados; en Estados Unidos se ha triplicado en los últimos 50 años.
Etiología
La etiología del cáncer pancreático se desconoce. Edad, tabaquismo y pancreatitis crónica se identifican como factores de riesgo. El cáncer pancreático rara vez se presenta en personas menores de 50 años, y el riesgo se incrementa con la edad. El factor de riesgo ambiental más significativo y reproducible es el tabaquismo. La incidencia de la enfermedad es del doble en fumadores, en comparación con no fumadores. La diabetes y la pancreatitis crónica también se relacionan con el cáncer pancreático; no obstante, ni la naturaleza ni la secuencia de la relación causa-efecto potencial se han establecido. La pancreatitis hereditaria y el síndrome familiar de mola atípica-melanoma múltiple son otras 2 afecciones vinculadas con este cáncer.
Manifestaciones clínicas
Casi todos los cánceres pancreáticos son adenocarcinomas del epitelio ductal, y sus síntomas se deben de manera primordial al efecto de masa más que a la pérdida de laintegridad de la función exocrina o endocrina. Las manifestaciones clínicas dependen del tamaño y la ubicación del tumor, y también de la presencia de metástasis. Dolor, ictericia y pérdida ponderal constituyen la manifestación clínica clásica del trastorno. El más frecuente es el dolor tipo sordo en el epigastrio, con frecuencia acompañado por dolor en la espalda, que muchas veces se intensifica en la posición supina y se alivia al inclinar el torso hacia adelante. Aunque el tumor puede originarse en cualquier sitio del páncreas, el punto de origen más frecuente es la cabeza (60%), a la que siguen el cuerpo (10%) y la cola (5%).
El páncreas muestra afectación difusa en el 25% de los casos restantes. Dada la proximidad del páncreas al conducto biliar común y el ámpula hepatopancreática, el cáncer de la cabeza del páncreas tiende a obstruir el flujo biliar. Con frecuencia la ictericia es el síntoma de presentación en la persona con cáncer de la cabeza del páncreas, y suele ir acompañada por referencia de dolor y prurito. El cáncer del cuerpo del páncreas por lo regular afecta al ganglio celíaco, por lo que produce dolor, que a menudo se intensifica tras la ingestión de alimentos o al asumir la posición supina. El cáncer de la cola del páncreas por lo general presenta metástasis antes de inducir síntomas.
La tromboflebitis migratoria (trombosis venosa profunda) se desarrolla en cerca del 10% de las personas con cáncer pancreático, en particular cuando el tumor afecta el cuerpo o la cola del páncreas. Los trombos se desarrollan en numerosas venas, entre otras, las venas profundas de las piernas, la vena subclavia, las venas mesentéricas inferior y superior, e incluso la vena cava. No es raro que la tromboflebitis migratoria constituya la primera evidencia del cáncer pancreático, aunque pudiera presentarse también en otros cánceres. El mecanismo responsable del estado de hipercoagulabilidad se desconoce en gran medida, pero pudiera vincularse con la activación de los factores de la coagulación debida a la acción de las proteasas liberadas a partir de las células tumorales.
Diagnóstico y tratamiento
El interrogatorio clínico, la exploración física y la elevación en las concentraciones séricas de bilirrubina y fosfatasa alcalina pueden sugerir la presencia del cáncer pancreático, pero no son diagnósticos. El antígeno del cáncer (AC) 19-9 en el suero, un antígeno del grupo sanguíneo de Lewis, puede ser útil para confirmar el diagnóstico en pacientes sintomáticos, y ayudar a establecer el pronóstico y la recurrencia tras la resección. Sin embargo, el AC 19-9 tiene una sensibilidad y una especificidad del 80% al 90%, de tal manera que no confirma el diagnóstico.
La ecografía y los estudios de TC son las técnicas de empleo más frecuente para confirmar el diagnóstico. La técnica preferida para obtener imágenes del páncreas es la TC espiral reforzada con contraste intravenoso y oral. La aspiración percutánea con aguja fina para realizar un estudio citológico del páncreas ha constituido uno de los principales avances para el diagnóstico del cáncer pancreático. Desafortunadamente, es bastante probable que esta técnica sea incapaz de detectar los tumores más pequeños y sensibles de curación. Es posible recurrir a la CPRE para evaluar a los pacientes con sospecha de cáncer pancreático e ictericia obstructiva.
La resección quirúrgica del tumor se realiza cuando éste se encuentra localizado. Sin embargo, esto sólo tiene lugar en un 10% a un 15% de las personas debido a que casi todos los cánceres del páncreas tienen ya metástasis en el momento del diagnóstico. En otro sentido, la resección quirúrgica se reserva como medida paliativa. Es posible que la radioterapia resulte útil cuando la enfermedad no es resecable pero parece encontrarse localizada. El empleo de radioterapia y quimioterapia para el cáncer de páncreas sigue en investigación. El control del dolor es uno de los aspectos más importantes del manejo en pacientes con cáncer pancreático en fase terminal.