01. Hígado y sistema hepatobiliar
El hígado es el órgano visceral más grande del cuerpo; pesa alrededor de 1,3 kg en el adulto. Se ubica por debajo del diafragma y ocupa gran parte del hipocondrio derecho (figura 46-1).
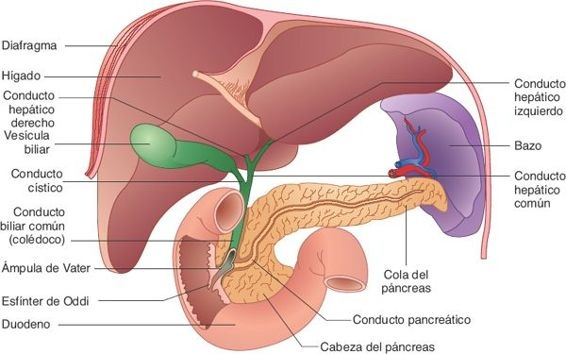
Una cápsula fibroelástica resistente, denominada cápsula de Glisson, lo circunda. Desde la perspectiva anatómica, el hígado se divide en 2 lóbulos grandes (lóbulos derecho e izquierdo) y 2 lóbulos más pequeños (los lóbulos caudado y cuadrado). Excepto por la porción que se ubica en el área epigástrica, el hígado se encuentra contenido por la caja torácica y, por lo regular, no puede palparse en personas saludables.
El hígado recibe el 25% del gasto cardíaco en reposo. Entre los órganos abdominales es el único que cuenta con una irrigación sanguínea doble, constituida por una irrigación venosa (portal) que llega por la vena porta hepática, y una irrigación arterial derivada de la arteria hepática.
Alrededor del 25% de la sangre que ingresa cada minuto al hígado llega por la arteria hepática; el 75% restante ingresa por medio de la vena porta, que carece de válvulas. La sangre venosa que llega por la vena porta hepática proviene del tubo digestivo y de los órganos abdominales principales, entre otros, el páncreas y el bazo (figura 46-2).
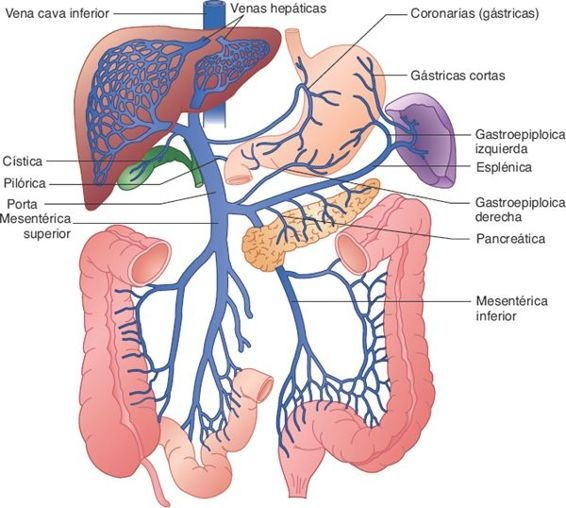
La circulación sanguínea portal lleva nutrimentos y materiales tóxicos que se absorben en el intestino, células hemáticas y sus productos de degradación a partir del bazo e insulina y glucagón del páncreas. Si bien la sangre de la vena porta muestra una saturación incompleta de oxígeno, cubre alrededor del 75% de los requerimientos de oxígeno del hígado.
El flujo venoso de salida del hígado tiene lugar a través de las venas hepáticas que carecen de válvulas y drenan en la vena cava inferior justo por debajo del diafragma. A menudo la diferencia de presión entre la vena hepática y la vena porta es tal, que el hígado tiene capacidad para almacenar alrededor de 500 ml a 1.000 ml de sangre. Esta sangre puede movilizarse hacia la circulación general durante los períodos de hipovolemia y shock. En la insuficiencia cardíaca derecha, cuando la presión en la vena cava aumenta, la sangre retrocede y se acumula en el hígado.
Los lobulillos son las unidades funcionales del hígado. Cada uno es una estructura cilíndrica que mide alrededor de 0,8 mm a 2 mm de diámetro, y varios milímetros de longitud. Existen alrededor de 50.000 a 100.000 lobulillos en el hígado. Cada lobulillo se organiza en torno a una vena central que drena en las venas hepáticas, drenar es correcto y de ahí en la vena cava. Los conductos biliares terminales y las ramificaciones pequeñas de la vena porta y la arteria hepática se ubican en la periferia de lobulillos. Placas de células hepáticas irradian en sentido centrífugo a partir de la vena central, como los radios de una rueda (figura 46-3). Estas placas hepáticas se encuentran separadas por capilares sinusoidales anchos, de pared delgada, denominado sinusoides, que se extienden de la periferia de lobulillos hacia su vena central.
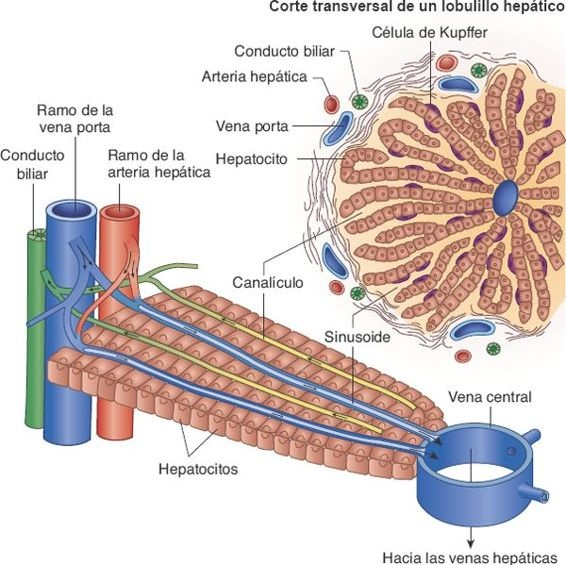
Los sinusoides reciben irrigación de la sangre de la vena porta y de la arteria hepática. Se encuentran en contacto íntimo con las células hepáticas, y participan en el intercambio de sustancias entre la sangre y los hepatocitos. Están recubiertos con 2 tipos de células: las células endoteliales capilares típicas y las células de Kupffer. Estas últimas son células reticuloendoteliales capaces de eliminar y fagocitar células hemáticas viejas y defectuosas, bacterias y otros materiales extraños que llegan en la sangre portal al tiempo que fluyen por el sinusoide. Esta acción fagocítica elimina los bacilos entéricos y otras sustancias lesivas que se filtran hacia la sangre a partir del intestino.
Una importante función exocrina del hígado es la secreción de bilis. Pequeños canales tubulares, denominados canalículos biliares, ubicados entre las membranas celulares de los hepatocitos adyacentes, también suplen a los lóbulos. La bilis que producen los hepatocitos fluye hacia los canalículos y luego hacia la periferia de los lobulillos, para drenar en conductos cada vez mayores, hasta que llegan a los conductos hepáticos derecho e izquierdo. Con frecuencia, los conductos biliares intrahepáticos y extrahepáticos se denominan, en conjunto, árbol hepatobiliar. Estos conductos se unen para constituir el conducto biliar común, que tiene alrededor de 10 cm a 15 cm de longitud, se orienta en dirección caudal, pasa por detrás del páncreas e ingresa al duodeno. El conducto pancreático se une al conducto biliar común en un conducto corto dilatado llamado ámpula hepatopancreática (ámpula de Vater), que drena en el duodeno a través de la papila duodenal. El tejido muscular en la unión de la papila, que en ocasiones se denomina esfínter de Oddi, regula el flujo de bilis hacia el duodeno. Cuando este esfínter se encuentra cerrado, la bilis se desplaza en dirección retrógrada hacia el conducto biliar común y la vesícula biliar.
Funciones metabólicas del hígado
El hígado es uno de los órganos más versátiles y activos del organismo. Produce la bilis, metaboliza hormonas y fármacos; sintetiza proteínas, glucosa y factores de la coagulación; almacena vitaminas y minerales; transforma en urea el amoníaco que se produce por la desaminación de los aminoácidos,y convierte los ácidos grasos en cetonas. También degrada los nutrimentos excedentes y los convierte en sustancias esenciales para el organismo. En su capacidad para metabolizar los fármacos y las hormonas, el hígado funge como un órgano excretor. En este sentido, la bilis, que transporta los productos finales de las sustancias que se metabolizan en el hígado, es en gran medida como la orina, que lleva los desechos corporales que filtran los riñones. Las funciones del hígado se resumen en la tabla 46-1.
Metabolismo de carbohidratos
El hígado desempeña un papel esencial en el metabolismo de los carbohidratos y la homeostasis de la glucosa (figura 46-4).
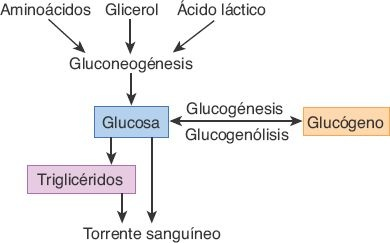
Los hepatocitos tienen la capacidad de almacenar grandes cantidades de glucosa en forma de glucógeno, mediante un proceso denominado glucogénesis. Cuando las concentraciones de glucosa en la sangre son bajas, el glucógeno vuelve a convertirse en glucosa por medio de la glucogenólisis, proceso en que participa una enzima fosfatasa específica de las células hepáticas. El hígado también produce glucosa a partir de los aminoácidos, el glicerol y el ácido láctico, como medio para mantener la glucemia durante los períodos de ayuno o incremento de la demanda. El hígado, además, convierte el exceso de carbohidratos en triglicéridos, para permitir su almacenamiento en el tejido adiposo.
Síntesis de proteínas y conversión del amoníaco en urea
El hígado es un sitio importante para la síntesis y degradación de las proteínas. Produce las proteínas para cubrir sus propios requerimientos celulares, y también las proteínas secretoras que se liberan hacia la circulación. La albúmina es la más importante de las proteínas secretoras; contribuye de manera significativa a la presión coloidosmótica del plasma, así como a la unión y el trasporte de numerosas sustancias, entre éstas, algunas hormonas, ácidos grasos, bilirrubina y otros aniones. El hígado produce asimismo otras proteínas importantes, como el fibrinógeno y los factores de la coagulación.
Mediante distintos procesos anabólicos y catabólicos, el hígado es el sitio principal para la interconversión de aminoácidos (figura 46-5).
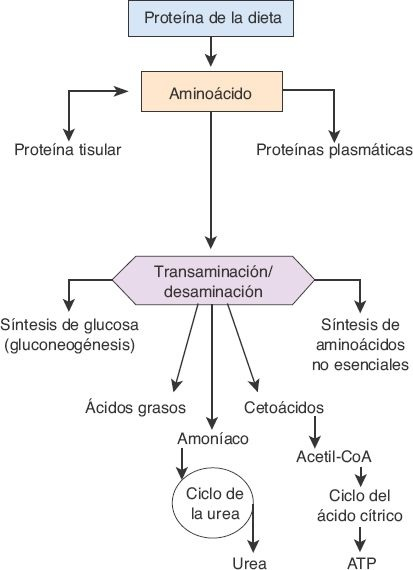
El catabolismo y la degradación en el hígado implican 2 reacciones básicas: la transaminación y la desaminación. En la transaminación se transfiere un grupo amino (NH2) a una sustancia aceptora. Como consecuencia de la transaminación, los aminoácidos pueden participar en el metabolismo intermedio de los carbohidratos y los lípidos.
Durante los períodos de ayuno o inanición, los aminoácidos se utilizan en la producción de glucosa (es decir, la gluconeogénesis). La mayor parte de los aminoácidos no esenciales se sintetiza en el hígado mediante transaminación, un proceso que es catalizado por las aminotransferasas, enzimas que se encuentran en grandes cantidades en este órgano.
La desaminación oxidativa implica el retiro de grupos amino a partir de los aminoácidos, y la conversión de estos últimos en cetoácidos y amoníaco. Esto sucede sobre todo mediante transaminación, en la que se retiran grupos amino y luego se transfieren a otra sustancia aceptora que, entonces, puede trasladar el grupo amino a otra sustancia más, o liberarlo en forma de amoníaco. Dado que el amoníaco es muy tóxico para los tejidos corporales, en particular para las neuronas, el que se libera durante la desaminación se retira de la sangre en el hígado con rapidez y se convierte en urea. En esencia, toda la urea que se forma en el organismo se obtiene por medio del ciclo de la urea en el hígado, y luego se excreta, sobre todo, a través de los riñones. Cierta cantidad se difunde hacia el intestino, donde se convierte en amoníaco por la acción de las bacterias entéricas.
La producción intestinal de amoníaco también deriva de la desaminación bacteriana de los aminoácidos y las proteínas de la dieta que no se absorben, de las células exfoliadas o de la sangre en el tubo gastrointestinal. Este amoníaco pasa a la circulación portal y se transporta hacia el hígado, donde se convierte en urea antes de alcanzar la circulación sistémica. La producción intestinal de amoníaco se incrementa después de la ingestión de alimentos ricos en proteínas y con la hemorragia gastrointestinal. En la hepatopatía avanzada es frecuente el compromiso de la síntesis de urea, lo que origina la acumulación del amoníaco en la sangre.
Vías para el metabolismo lipídico
Si bien la mayoría de las células del organismo metaboliza los lípidos, algunos aspectos de este metabolismo tienen lugar sobre todo en el hígado, lo que incluye la oxidación de los ácidos grasos libres para obtener cetoácidos que aportan energía para otras funciones corporales; la síntesis del colesterol, los fosfolípidos y las lipoproteínas, y la formación de triglicéridos a partir de carbohidratos y proteínas (figura 46-6).
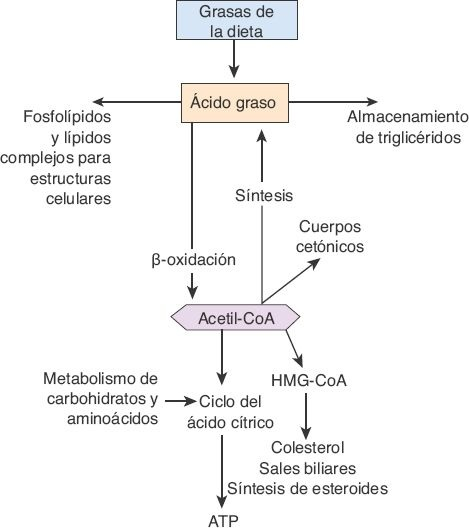
Para obtener energía a partir de los triglicéridos, la molécula debe, primero, dividirse en glicerol y ácidos grasos, para luego escindir a los ácidos grasos en unidades de 2 carbonos de acetilcoenzima A (acetil-CoA) mediante un proceso denominado β-oxidación. El acetil-CoA se canaliza con facilidad hacia el ciclo del ácido cítrico para generar trifosfato de adenosina (ATP). Dado que el hígado no puede utilizar todo el acetil-CoA que se forma, convierte el exceso en ácido acetoacético, un cetoácido muy soluble que se libera hacia el torrente sanguíneo y se transporta hacia otros tejidos, donde se usa para obtener energía. Durante los períodos de inanición, las cetonas se convierten en una fuente importante de energía, al tiempo que los ácidos grasos liberados a partir del tejido adiposo se convierten en cetonas en el hígado.
Las unidades de acetil-CoA derivadas del metabolismo de las grasas también se emplean para sintetizar colesterol y ácidos biliares en el hígado. En este órgano, el colesterol puede utilizarse en distintas formas: puede esterificarse y almacenarse; puede exportarse unido a lipoproteínas, o puede convertirse en ácidos biliares. El paso limitante de la velocidad para la síntesis del colesterol es el que cataliza la reductasa de la 3-hidroxi-3-metil-glutaril-coenzima A (reductasa de la HMG-CoA).
Los inhibidores de la reductasa de la HMG-CoA, o estatinas (fluvastatina, lovastatina, pravastatina y atorvastatina), se utilizan para controlar las concentraciones elevadas de colesterol, al inhibir este paso en la síntesis de la sustancia.
En el organismo, casi toda la síntesis de lípidos a partir de carbohidratos y proteínas tiene lugar en el hígado. Así, siempre que ingresa una cantidad de carbohidratos mayor a la que puede utilizarse de inmediato, el exceso se convierte en triglicéridos en el hígado. Estos triglicéridos se transportan sobre todo en lipoproteínas de baja densidad (LBD) hacia el tejido adiposo, donde se almacenan.
Producción de bilis y colestasis
La secreción de bilis es esencial para la digestión de las grasas de la dieta y la absorción de éstas y de las vitaminas liposolubles a partir del intestino. El hígado produce a diario alrededor de 500 ml a 600 ml de bilis de color amarillo-verdoso. La bilis contiene agua, sales biliares, bilirrubina, colesterol y ciertos productos colaterales del metabolismo. De éstos, sólo las sales biliares, que se forman a partir del colesterol, son importantes para la digestión. Los otros componentes dependen de la secreción de sodio, cloruro, bicarbonato y potasio en los conductos biliares.
Las sales biliares tienen un papel importante en la digestión; facilitan la emulsificación de las grasas de la dieta, y son necesarias para la formación de los micelios que transportan a los ácidos grasos y a las vitaminas liposolubles hacia la superficie de la mucosa intestinal para su absorción. El sistema para la recirculación de la bilis, la circulación enterohepática, incluye componentes múltiples. El hígado, el árbol biliar, la vesícula biliar, la circulación venosa portal, el intestino delgado, el colon y los riñones, desempeñan algún papel en grado variable. Más del 90% de las sales biliares que ingresan al intestino se reabsorbe hacia la circulación portal por medio de un proceso de transporte activo que tiene lugar en la región distal del íleon. A partir de la circulación portal, las sales biliares se desplazan hacia el interior de las células hepáticas, y se reciclan. Por lo general, las sales biliares pasan por todo este circuito alrededor de 17 veces antes de ser expulsadas en las heces.
Colestasis
La colestasis representa una disminución del flujo biliar por los canalículos intrahepáticos, y una reducción en la secreción de agua, bilirrubina y ácidos biliares a partir de los hepatocitos. Como consecuencia, los materiales que por lo regular se transfieren hacia la bilis, entre otros la bilirrubina, el colesterol y los ácidos biliares, se acumulan en la sangre. La condición puede derivar de una hepatopatía intrínseca, en cuyo caso se denomina colestasis intrahepática, o de la obstrucción de los conductos biliares mayores, que origina lo que se conoce como colestasis extrahepática.
Distintos mecanismos están implicados en la patogénesis de la colestasis. La cirrosis biliar primaria (un trastorno autoinmunitario) y la colangitis esclerosante primaria se deben a trastornos de los canalículos intrahepáticos pequeños y los conductos biliares. En el caso de la obstrucción extrahepática, que puede derivar de afecciones como la colelitiasis, las estenosis del conducto biliar común o de las neoplasias que generan obstrucción, los efectos comienzan con un incremento de la presión dentro de los conductos biliares grandes. Los trastornos genéticos que pueden producir colestasis incluyen la colestasis benigna recurrente, el síndrome de Byler y el síndrome de Alagille.
La colestasis benigna recurrente afecta al transporte de bilis hacia los canalículos. El síndrome de Byler también se conoce como colestasis intrahepática familiar progresiva tipo I. La mutación genética responsable de la enfermedad induce diarrea, prurito e insuficiencia hepática. El gen también se ubica en el intestino delgado y el páncreas, y afecta los sistemas gastrointestinal y endocrino. El síndrome de Alagille es un trastorno autosómico dominante que da origen a una hipoplasia intrahepática que afecta de manera específica los conductos biliares interlobares. Los pacientes con el síndrome presentan anomalías cardíacas y oftálmicas aunadas a malformaciones esqueléticas, de manera específica en los huesos faciales.
Las características morfológicas de la colestasis varían según la causa subyacente. La acumulación de pigmento biliar en el hígado es común a todos los tipos de colestasis obstructiva y hepatocelular. En los canalículos biliares dilatados pueden observarse tapones elongados de bilis de color verde-pardo. La ruptura de los canalículos conduce a la extravasación de la bilis y a cambios degenerativos subsecuentes en los hepatocitos circundantes. La colestasis obstructiva prolongada no sólo determina cambios grasos en los hepatocitos, sino la destrucción del tejido conectivo de soporte, lo que da origen a reservorios de bilis que contienen de tritos celulares y pigmento. La obstrucción no resuelta conduce a la fibrosis de las vías biliares y, por último, a la cirrosis biliar en fase terminal.
El prurito es el síntoma más frecuente en las personas con colestasis, y quizá esté relacionado con un incremento de los ácidos biliares en el plasma. Pueden desarrollarse xantomas cutáneos (acumulaciones localizadas de colesterol), resultado de la hiperlipidemia y de las anomalías para la excreción del colesterol. Un hallazgo de laboratorio característico es la elevación en la concentración sérica de fosfatasa alcalina, una enzima presente en el epitelio del conducto biliar y la membrana canalicular de los hepatocitos. Otras manifestaciones de la disminución del flujo biliar se asocian con la absorción intestinal, e incluyen insuficiencias nutricionales de las vitaminas liposolubles A, D y K.
Eliminación de bilirrubina e ictericia
La bilirrubina es el producto final de la degradación del grupo hem que contienen los eritrocitos viejos. Es la sustancia que confiere a la bilis su color. En el proceso de degradación, la hemoglobina que deriva del eritrocito se degrada para formar biliverdina, que se convierte con rapidez en bilirrubina libre (figura 46-7). Ésta, que es insoluble en el plasma, se transporta en la sangre unida a la albúmina plasmática, pero incluso cuando se da esta unión se sigue nombrando bilirrubina libre, para distinguirla de la bilirrubina conjugada. Al tiempo que pasa por el hígado, la bilirrubina libre se traslada a través de la membrana de los hepatocitos y se libera de su molécula de albúmina portadora.
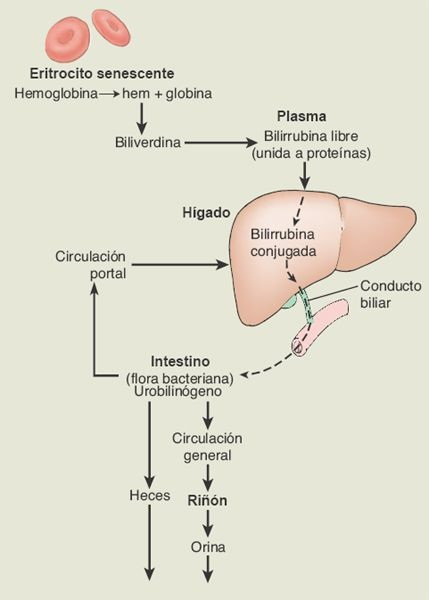
Dentro de los hepatocitos, la bilirrubina libre se convierte en bilirrubina conjugada, lo que la hace soluble en la bilis. La bilirrubina conjugada se excreta como un constituyente de la bilis, y en esta forma pasa por los conductos biliares hacia el intestino delgado donde, debido a la acción de la flora intestinal, cerca de la mitad de la bilirrubina se convierte en una sustancia muy soluble denominada urobilinógeno. Alrededor de una quinta parte del urobilinógeno que se produce se absorbe hacia la circulación portal, y el remanente se excreta en las heces. La mayor parte del urobilinógeno que se absorbe regresa al hígado para volver a excretarse hacia la bilis.
Por lo general, en la sangre sólo se detecta una cantidad escasa de bilirrubina; la concentración normal de bilirrubina sérica total es menor de 1,5 mg/dl (17 μmol a 20,5 μmol). Las mediciones de bilirrubina realizadas en el laboratorio suelen cuantificar tanto la bilirrubina libre como la conjugada, y también la bilirrubina total. Estos resultados se informan como bilirrubina directa (conjugada) y bilirrubina indirecta (no conjugada o libre).
Ictericia
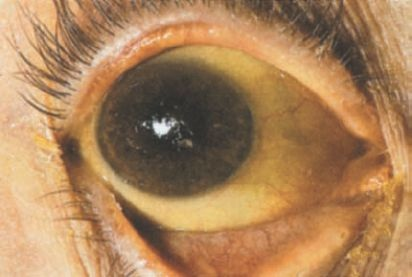
La ictericia, o la pigmentación amarillenta de la piel y los tejidos profundos, deriva de las concentraciones elevadas anómalas de bilirrubina en la sangre. Se desarrolla cuando existe un desequilibrio entre la síntesis de bilirrubina y su eliminación. La ictericia se hace evidente cuando las concentraciones de bilirrubina sérica se elevan por encima de 2 mg/dl a 2,5 mg/dl (34,2 μmol a 42,8 μmol) 5,10 . Debido a que la piel normal tiene un tono amarillo, los signos tempranos de la ictericia son muchas veces difíciles de detectar, en particular en personas de piel oscura. La bilirrubina tiene afinidad singular por el tejido elástico. La esclerótica del ojo, que contiene una proporción alta de fibras elásticas, suele ser una de las primeras estructuras en que puede detectarse la ictericia (figura 46-8).
Las 5 causas principales de ictericia son la destrucción excesiva de eritrocitos, las anomalías en la captación de la bilirrubina en los hepatocitos, la disminución de la conjugación de la bilirrubina, la obstrucción al flujo biliar en los canalículos de los lobulillos hepáticos o en los conductos biliares intrahepáticos o extrahepáticos, y la excesiva síntesis extrahepática de bilirrubina. Desde el punto de vista anatómico, la ictericia puede clasificarse como prehepática, intrahepática y poshepática. El cuadro 46-1 enumera las causas comunes de estos tipos de ictericia.
La causa primordial de ictericia prehepática es la hemólisis excesiva de los eritrocitos. La ictericia hemolítica se desarrolla cuando los eritrocitos se destruyen a una velocidad que excede la capacidad del hígado para eliminar la bilirrubina de la sangre. Puede presentarse tras una reacción hemolítica posterior a una transfusión sanguínea, por una disminución del período de vida de los eritrocitos donados, o en trastornos como la esferocitosis hereditaria, en la que las membranas eritrocitarias son defectuosas, o bien en la enfermedad hemolítica del recién nacido. Cuando se presenta una hemorragia interna también puede existir una producción excesiva de bilirrubina al tiempo que tiene lugar la reabsorción de la sangre. Además, los trastornos que generan una eritropoyesis ineficaz pueden incrementar la generación de bilirrubina. La hiperbilirrubinemia neonatal se debe a un aumento en la producción de bilirrubina en los recién nacidos y a su capacidad limitada para excretarla entre los días 0 y 14 de vida. Los neonatos prematuros se encuentran en un riesgo particu lar debido a que sus eritrocitos tienen una vida más corta y una tasa de recambio mayor. En la ictericia prehepática el trastorno pigmentario es leve, se eleva la bilirrubina no conjugada, las heces tienen coloración normal y no se detecta bilirrubina en la orina.
La ictericia intrahepática o hepatocelular se debe a trastornos que afectan de manera directa la capacidad del hígado para retirar la bilirrubina de la sangre o conjugarla, de tal forma que pueda ser eliminada en la bilis. La enfermedad de Gilbert se hereda como rasgo dominante y provoca una disminución, del 66% en promedio, del retiro de la bilirrubina de la sangre. El trastorno es benigno y bastante común, con una tasa de prevalencia aproximada del 8%. Las personas afectadas son asintomáticas, excepto por una concentración un poco elevada de bilirrubina no conjugada e ictericia leve. La conjugación de la bilirrubina se encuentra comprometida siempre que las células hepáticas se dañan, cuando el trasporte de bilirrubina hacia el interior de los hepatocitos se vuelve deficiente o cuando se carece de las enzimas necesarias para conjugar la bilis. Las hepatopatías, como la hepatitis y la cirrosis, son las causas más frecuentes de ictericia intrahepática. Los fármacos, como el anestésico halotano, anticonceptivos orales, estrógenos, esteroides anabólicos, isoniazida,rifampicina y clorpromazina también pueden estar implicados en este tipo de ictericia. La ictericia intrahepática o hepatocelular suele interferir con todas las fases del metabolismo de la bilirrubina: captación, conjugación y excreción. Tanto la bilirrubina conjugada como la no conjugada se encuentran elevadas; con frecuencia la orina es oscura debido a la presencia de bilirrubina en ella, y la fosfatasa alcalina sérica muestra un aumento ligero.
La ictericia poshepática u obstructiva, también denominada ictericia colestásica, tiene lugar cuando existe obstrucción al flujo biliar entre el hígado y el intestino; la obstrucción podría ubicarse en cualquier punto entre la unión del conducto hepático derecho o izquierdo y el sitio en que el conducto biliar se abre en el intestino. Entre las causas se encuentran la estenosis del conducto biliar, los litos vesiculares y los tumores del conducto biliar o del páncreas. La bilirrubina conjugada suele mostrar elevación; las heces tienen color blanquecino debido a la carencia de bilirrubina en la bilis; la orina es oscura; las concentraciones séricas de fosfatasa alcalina muestran incremento marcado, y los niveles de aminotransferasa muestran aumento discreto. Las concentraciones sanguíneas de ácidos biliares suelen ser altas en la ictericia obstructiva. Al tiempo que los ácidos biliares se acumulan en la sangre, se desarrolla prurito. El prurito que precede a la ictericia es común en la ictericia obstructiva.
Pruebas de función hepatobiliar
En la mayoría de los casos, la anamnesis y exploración física aportan información clave sobre la función hepática. Las pruebas diagnósticas ayudan a evaluar la actividad del hígado y el grado de daño que presenta el órgano. Las pruebas de laboratorio se utilizan con frecuencia para valorar la función hepática y confirmar el diagnóstico de hepatopatía.
Las pruebas de función hepática, que incluyen las concentraciones séricas de enzimas hepáticas, se usan para facilitar el diagnóstico de la enfermedad, diferenciar entre los distintos trastornos, determinar la gravedad del padecimiento existente y vigilar las respuestas al tratamiento establecido. Los valores elevados en las pruebas de enzimas séricas suelen indicar la existencia de lesión hepática antes que otros indicadores de la función hepática. Las enzimas clave son la aminotransferasa de alanina (ATL) y la aminotransferasa de aspartato (ATS), que se encuentran en todos los hepatocitos. La ATL es específica del hígado, en tanto que la ATS deriva de órganos distintos al hígado. En la mayor parte de los casos de daño hepático existen incrementos paralelos de la ATL y la ATS. El aumento más radical se observa en los casos de lesión hepatocelular aguda, como sucede en la hepatitis viral, la lesión hipóxica o isquémica, la lesión tóxica aguda o el síndrome de Reye.
La capacidad de síntesis del hígado se refleja en las mediciones de las concentraciones de proteínas séricas y el tiempo de protrombina (es decir, síntesis de factores de la coagulación). La hipoalbuminemia secundaria a la depresión de la síntesis puede complicar la hepatopatía grave. Suelen presentarse insuficiencias de los factores de la coagulación V y los dependientes de la vitamina K (II, VII, IX y X).
La bilirrubina sérica, la gammaglutamiltransferasa (GGT), la 5’-nucleotidasa y la fosfatasa alcalina miden la función excretora del hígado. La fosfatasa alcalina y la 5’-nucleotidasa se identifican en las membranas ubicadas entre los hepatocitos y el conducto biliar, y son liberadas por trastornos que afectan al conducto biliar. La GGT se localiza en el retículo endoplásmico de los hepatocitos y en las células epiteliales del conducto biliar. Se cree que participa en el transporte de los aminoácidos y los péptidos hacia el interior de los hepatocitos. La cuantificación de la GGT puede resultar útil para el diagnóstico de consumo excesivo de alcohol y es un indicador de enfermedad hepatobiliar.
La ecografía aporta información sobre el tamaño, la composición y el flujo sanguíneo del hígado. Ha sustituido en gran medida a la colangiografía para la detección de litos en la vesícula biliar o el árbol biliar. La tomografía computarizada (TC) aporta información similar a la que se obtiene con la ecografía. Las imágenes por resonancia magnética (IRM) han probado ser útiles en algunos trastornos. La angiografía selectiva de las arterias celíaca, mesentérica superior o hepática, puede utilizarse para visualizar la circulación hepática o portal. Una biopsia hepática constituye un medio para analizar el tejido hepático sin cirugía. Existen varias técnicas para obtener tejido hepático: biopsia hepática percutánea, que recurre a una aguja para succión, corte, o corte de carga con resorte; biopsia hepática laparoscópica, y biopsia con aguja fina, que se lleva a cabo con guía ultrasonográfica o con TC. El método a utilizar depende del número de especímenes que se requiere y de la cantidad de tejido necesario para la evaluación. La biopsia hepática laparoscópica constituye un medio para examinar las masas abdominales, evaluar la ascitis de causa desconocida y realizar el estadiaje de los cánceres hepáticos.