04. Trastornos de la función glomerular
Los glomérulos son ovillos de capilares que yacen entre las arteriolas aferentes y eferentes. Los capilares de los glomérulos están dispuestos en lóbulos y apoyados por un tallo que consiste en células mesangiales y una matriz basal parecida a una membrana (figura 41-9).
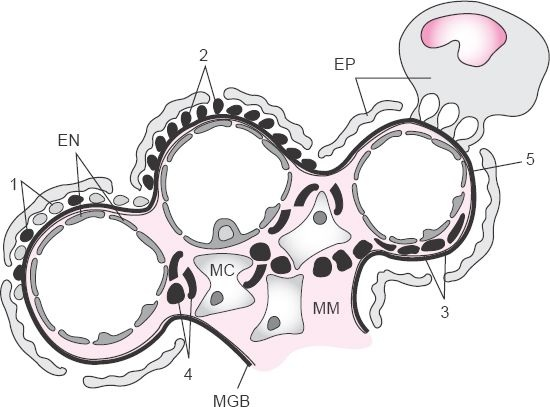
La membrana capilar glomerular está compuesta de 3 capas estructurales: una capa de células endoteliales que reviste la superficie interna del capilar, una membrana basal constituida de una red de proteínas matriciales y una capa de células epiteliales que rodea a la superficie externa del capilar y reviste la superficie interna de la cápsula de Bowman. Las células endoteliales están unidas a la membrana basal por largos procesos parecidos a pies (podocitos) que encierran la membrana externa de los capilares. La membrana capilar glomerular es selectivamente permeable, permitiendo que el agua y partículas pequeñas (ej. electrolitos y partículas disueltas, tales como glucosa y aminoácidos) salgan de la sangre y entren al espacio de Bowman y evitando que partículas más grandes (ej. proteínas plasmáticas y células sanguíneas) salgan de la sangre.
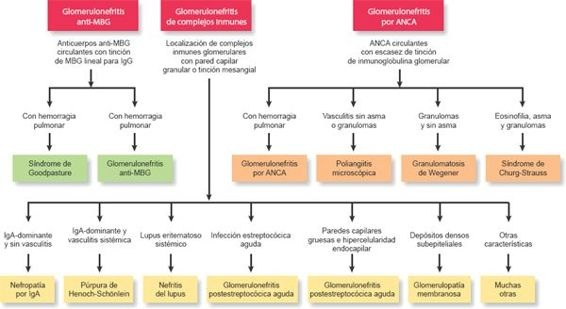
La glomerulonefritis, un proceso inflamatorio que afecta a estructuras glomerulares, es la segunda causa principal de insuficiencia renal a nivel mundial y se clasifica en tercer lugar, después de la diabetes y la hipertensión, como causa de enfermedad renal crónica en Estados Unidos. Hay muchos casos de enfermedad glomerular. La enfermedad podría ocurrir como una afección primaria en la que la anomalía glomerular es la única enfermedad presente o podría ocurrir como una afección secundaria en la que la anomalía glomerular resulta de otra enfermedad, tal como diabetes mellitus. Véase en la figura 41-10 un algoritmo respecto a la glomerulonefritis primaria frente a secundaria.
Causas y patogénesis de la lesión glomerular
Los agentes causantes o eventos activadores que producen lesión glomerular incluyen mecanismos inmunitarios, no inmunitarios y hereditarios. La mayoría de casos primarios y muchos casos de enfermedad glomerular secundaria tienen probablemente un origen inmune. Aunque muchas enfermedades glomerulares son impulsadas por eventos inmunitarios, diversas tensiones metabólicas no inmunitarios (ej. diabetes), hemodinámicas (ej. hipertensión) y tóxicos (ej. fármacos y productos químicos) pueden inducir lesión glomerular, ya sea solos o junto con mecanismos inmunitarios. Enfermedades glomerulares hereditarias tales como el síndrome de Alport, aunque relativamente raro, son una categoría importante de enfermedad glomerular como resultado de su asociación con la pérdida progresiva de función y transmisión renal a futuras generaciones.
Dos tipos de mecanismos inmunes han sido implicados en el desarrollo de enfermedad glomerular:
- Lesión que resulta de anticuerpos que reaccionan con antígenos glomerulares fijos o antígenos plantados dentro del glomérulo.
- La lesión que resulta de complejos circulantes antígenoanticuerpo que quedan atrapados en la membrana glomerular (figura 41-11).
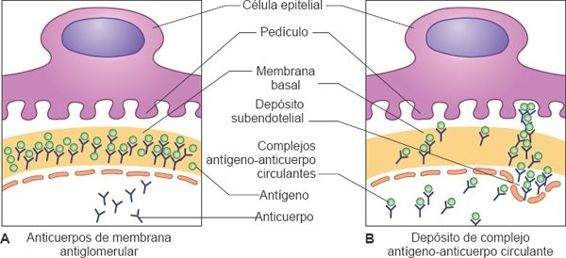
Los antígenos responsables del desarrollo de la respuesta inmune podrían ser de origen endógeno, tales como los anticuerpos para el ácido desoxirribonucleico (ADN) en el LES o podrían ser de origen exógeno, tales como los antígenos de la membrana estreptocócica en la glomerulonefritis postestreptocócica. Frecuentemente, se desconoce la fuente del antígeno.
Los cambios celulares que ocurren con la enfermedad glomerular incluyen incrementos en el número de células glomerulares o inflamatorias (proliferativas o hipercelulares), engrosamiento de la membrana basal (membranoso) y cambios en los componentes glomerulares no celulares (esclerosis y fibrosis). Un incremento en los números de células se caracteriza por uno o más de los hechos siguientes: proliferación de células endoteliales y mesangiales, infiltración leucocitaria (neutrófilos, monocitos y, en algunos casos, linfocitos) y formación de medias lunas (colecciones en forma de media luna de células epiteliales proliferantes y leucocitos infiltrantes) en el espacio de Bowman.
El engrosamiento de la membrana basal conlleva depósito de material no celular denso en los lados endotelial y epitelial de la membrana basal o dentro de la membrana misma. Esclerosis se refiere a un incremento en la cantidad de material extracelular en el tejido mesangial, subendotelial o subepitelial del glomérulo, y fibrosis se refiere al depósito de fibras de colágeno. Los cambios glomerulares pueden ser difusos, afectando los glomérulos y las partes del glomérulo; focales, en los que sólo algunos glomérulos son afectados y otros son esencialmente normales; segmentarios, afectando sólo un cierto segmento de cada glomérulo; o mesangiales, afectando sólo células mesangiales. En la figura 41-9B se ilustra la ubicación de las lesiones relacionadas con varios tipos de enfermedad glomerular.
Tipos de enfermedad glomerular
Las manifestaciones clínicas de los trastornos glomerulares caen, por lo general, en una de 5 categorías:
- Síndromes nefríticos.
- Glomerulonefritis rápidamente progresiva.
- Síndrome nefrótico.
- Trastornos asintomáticos del sedimento urinario (es decir, hematuria y proteinuria).
- Glomerulonefritis crónica.
Los síndromes nefríticos producen una respuesta inflamatoria proliferativa, mientras que el síndrome nefrótico produce permeabilidad incrementada del glomérulo. Debido a que la mayoría de trastornos glomerulares pueden producir síndromes nefríticos y nefróticos mixtos, un diagnóstico definitivo requiere con frecuencia biopsia renal.
Síndrome nefrítico agudo
El síndrome nefrítico agudo es la correlación clínica de la inflamación glomerular. En su forma más drástica, el síndrome nefrítico agudo se caracteriza por inicio repentino de hematuria (ya sea microscópica o extremadamente visible, con cilindros de glóbulos rojos), grados variables de proteinuria, TFG disminuida, oliguria y signos de función renal deteriorada. Lo causan los procesos inflamatorios que ocluyen el lumen capilar glomerular y el daño a la pared capilar. Este cambio a la pared capilar permite que los eritrocitos escapen hacia la orina y produzcan cambios hemodinámicos que disminuyen la TFG. La acumulación de líquido extracelular, hipertensión y edema se desarrollan como resultado de la TFG reducida e incrementan la reabsorción tubular de sal y agua.
El síndrome nefrítico agudo podría ocurrir en tales enfermedades sistémicas como LES. No obstante, se relaciona comúnmente con glomerulonefritis proliferativa aguda tal como la glo merulonefritis postinfecciosa.
Glomerulonefritis postinfecciosa aguda
La glomerulonefritis postinfecciosa aguda ocurre normalmente después de infección con ciertas cepas de estreptococos hemolíticos β del grupo A y es causada por el depósito de complejos inmunes de antígenos de anticuerpos y bacterianos. Podría ocurrir también después de infecciones por otros organismos, que incluyen estafilococos, un agente viral, tal como la hepatitis y varios parásitos. Aunque la enfermedad se observa principalmente en niños, personas de cualquier edad pueden ser afectadas.
La fase aguda de la glomerulonefritis postinfecciosa se caracteriza por agrandamiento glomerular difuso e hipercelularidad. La hipercelularidad es causada por infiltración de leucocitos, tanto neutrófilos como monocitos, y proliferación de células endoteliales y mesangiales. Hay también hinchazón de células endoteliales. La combinación de proliferación, hinchazón e infiltración leucocitaria destruye los lúmenes capilares glomerulares. Podría haber edema intersticial e inflamación, y los túbulos contienen con frecuencia eritrocitos. En las primeras semanas de la enfermedad, la microscopia de inmunofluorescencia revela, por lo general, depósitos granulares de IgG y el componente complemento C3 en el mesangio y a lo largo de la membrana basal (figura 41-12).
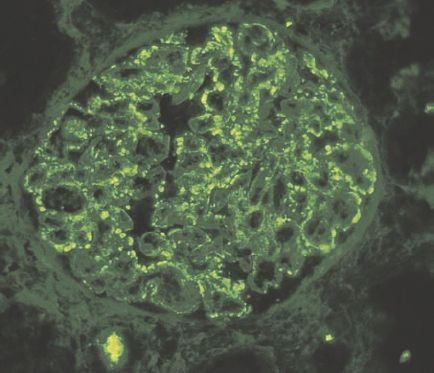
El caso clásico de la glomerulonefritis postestreptocócica sigue a una infección estreptocócica por aproximadamente 7 a 12 días. Este es el tiempo necesario para el desarrollo de anticuerpos. La infección primaria afecta por lo general a la faringe, pero podría ser activada por la piel. La oliguria, que se desarrolla cuando disminuye la TFG, es uno de los primeros síntomas. La proteinuria y hematuria siguen debido a la permeabilidad de la pared capilar glomerular. Los materiales en la orina degradan a los eritrocitos y la orina color cola podría el primer signo del trastorno. La retención de sodio y agua da lugar a edema (particularmente en cara y manos) e hipertensión. Los hallazgos de laboratorio importantes incluyen un anticuerpo antiestreptocócico elevado (AAE), una disminución de las concentraciones séricas de C3 y otros componentes de la cascada de complementos, y crioglobulinas (es decir, complejos inmunes grandes) en el suero.
El tratamiento de la glomerulonefritis postestreptocócica aguda incluye la eliminación de la infección estreptocócica con antibióticos y proporcionar atención de apoyo. El trastorno conlleva un pronóstico excelente y rara vez causa enfermedad renal crónica.
Glomerulonefritis rápidamente progresiva
La glomerulonefritis rápidamente progresiva es un síndrome caracterizado por signos y síntomas de lesión glomerular grave, que no tienen una causa específica. Coma indica su nombre, este tipo deglomerulonefritis es rápidamente progresiva, con frecuencia en cuestión de meses. El trastorno incluye la proliferación focal y segmentaria de células glomerulares y el reclutamiento de monocitos y macrófagos con formación de estructuras en forma de media luna que obstruyen el espacio de Bowman. La glomerulonefritis rápidamente progresiva podría ser causada por varios trastornos inmuitarios, algunos sistémicos y otros restringidos al riñón. Entre las enfermedades relacionadas con esta forma de glomerulonefritis están los trastornos complejos inmunes tales como el LES, vasculitis de vasos pequeños (ej. poliangitis microscópica) y un trastorno inmune llamado síndrome de Goodpasture.
Síndrome de Goodpasture
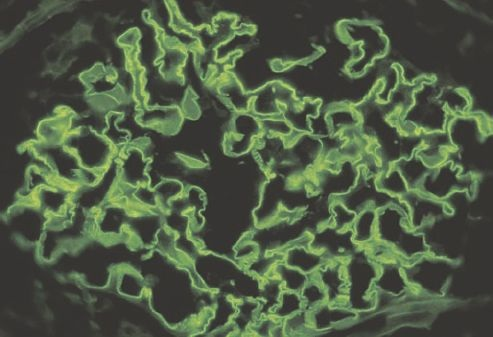
El síndrome de Goodpasture es una forma poco común y agresiva de la glomerulonefritis que es causada por anticuerpos de la membrana basal alveolar y glomerular (MBG). Los anticuerpos anti-MBG reaccionan transversalmente con la membrana basal alveolar pulmonar para producir el síndrome de hemorragia pulmonar relacionado con la insuficiencia renal.
El sello patológico de la glomerulonefritis anti-MBG es la tinción lineal difusa de las MBG para IgG (figura 41-13). Se desconoce la causa del trastorno, aunque la infección por influenza, exposición a disolventes de hidrocarburos (encontrados en pinturas y colorantes), varios fármacos y cáncer han sido relacionados en ciertas personas. Hay cierta tendencia a pensar que el síndrome de Goodpasture tiene una predisposición genética, pero no es concluyente.
El tratamiento incluye plasmaféresis para eliminar los anticuerpos anti-MBG y tratamiento inmunosupresor (es decir, corticosteroides y ciclofosfamida) para inhibir la producción de anticuerpos.
Síndrome nefrótico
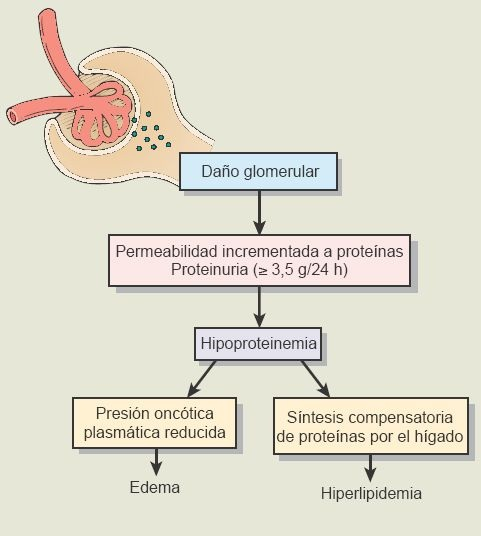
El síndrome nefrótico se caracteriza por proteinuria masiva (>3,5 g/día) y lipiduria (ej. grasa libre, cuerpos ovalados y cilindros adiposos) junto con una hipoalbuminemia relacionada (<3 g/dl), edema generalizado e hiperlipidemia (colesterol >300 mg/dl).El síndrome nefrótico no es una enfermedad glomerular específica, sino una constelación de hallazgos clínicos que resultan de un incremento en la permeabilidad glomerular y pérdida de proteínas plasmáticas en la orina(figura 41-14).
Patogénesis
Cualquier incremento en la permeabilidad de la membrana glomerular permite que las proteínas escapen del plasma hacia el filtrado glomerular. Resulta proteinuria masiva. que conduce a hipoalbuminemia. El edema generalizado, que es un sello del síndrome nefrótico, resulta de la pérdida de presión osmótica coloidal de la sangre con acumulación posterior de líquido en los tejidos intersticiales. Hay también retención de sal y agua, que agrave el edema. Esto, al parecer, se debe a varios factores, incluso un incremento compensatorio de aldosterona, estimulación del sistema nervioso simpático y una reducción de la secreción de factores natriuréticos. Al inicio, el edema se presente en partes dependientes del cuerpo tales como las extremidades inferiores, pero se vuelve generalizada cuando avanza la enfermedad. Disnea debido a edema pulmonar, efusiones pleurales y compromiso diafragmático debido a ascitis puede desarrollarse en personas con síndrome nefrítico.
La hiperlipidemia, que ocurre en personas con nefrosis, se caracteriza por concentraciones altas de triglicéridos y lipoproteínas de baja densidad (LBD). Las lipoproteínas de alta densidad (LAD) por lo común son normales. Debido a las concentraciones altas de LBD, las personas con síndrome nefrótico tienen mayor riesgo de desarrollar aterosclerosis.
La proporción más grande de proteína perdida en la orina es albúmina, pero podrían perderse también globulinas. Como resultado, las personas con nefrosis podrían ser vulnerables a infecciones, en particular las causadas por estafilococos y neumococos. Esta resistencia reducida a infección se relaciona probablemente con pérdida de inmunoglobulinas y componentes complemento de bajo peso molecular en la orina. Muchas proteínas de enlace se pierden también en la orina. En consecuencia, las concentraciones plasmáticas de muchos iones (hierro, cobre y cinc) y hormonas (hormonas tiroideas y sexuales) podrían ser bajas como resultado de la disminución de proteínas de enlace. Muchos fármacos requieren enlace de proteínas para transporte. La hipoalbuminemia reduce el número de sitios disponibles de enlace a proteína, produciendo así un posible incremento en la cantidad de fármaco libre (activo) que está disponible.
Causas
Los trastornos glomerulares que ocurren con la nefrosis pueden desarrollarse como un trastorno primario o secundario a cambios causados por enfermedades sistémicas tales como la diabetes mellitus y el LES. Entras las lesiones glomerulares primarias que conducen a síndrome nefrótico están la enfermedad de cambios mínimos (nefrosis lipoide), glomeruloesclerosis segmentaria focal y glomerulonefritis membranosa. La frecuencia relativa de estas causas varía con la edad. En niños mayores de 15 años de edad, el síndrome nefrótico casi siempre es causado por enfermedad glomerular idiopática primaria, mientras que en los adultos, es a menudo un trastorno secundario.
Enfermedad de cambios mínimos (nefrosis lipoide)
La enfermedad de cambios mínimos se caracteriza por pérdida difusa (a través de fusión) de los pedículos de células en la capa epitelial de la membrana glomerular. Es vista muy comúnmente en niños, pero podría ocurrir ocasionalmente en adultos. Se desconoce la causa de nefrosis de cambio mínimo. Aunque la enfermedad de cambios mínimos no avanza a insuficiencia renal, puede causar complicaciones significativas, incluso predisposición a infección con organismo grampositivos, una tendencia hacia eventos tromboembólicos, hiperlipidemia y desnutrición por proteínas.
Glomerulonefritis membranosa
La glomerulonefritis membranosa es la causa más común de nefrosis primaria en adultos, con mucha frecuencia en aquellos en las décadas quinta y sexta de vida y, casi siempre, después de la edad de 30 años. El trastorno es causado por engrosamiento difuso de la MBG debido a depósito de complejos inmunes. El trastorno podría ser idiopático o relacionado con varios trastornos, incluso enfermedades autoinmunes tales como el LES, infecciones tales como hepatitis B crónica y trastornos metabólicos tales como diabetes mellitus. La presencia de inmunoglobulinas y complemento en los depósitos subendoteliales sugiere que la enfermedad representa un trastorno crónico mediado por complejos inmunes.
El trastorno comienza, por lo general, con un inicio insidioso del síndrome nefrótico o, en un pequeño porcentaje de personas, con proteinuria no nefrótica. Hematuria e hipertensión leve podrían estar presentes. El avance de la enfermedad es variable. Algunas personas experimentan una remisión completa, otras tienen remisiones repetidas y recaídas, y todavía otras avanzan a insuficiencia renal completa e incluso la muerte. Las remisiones espontáneas y un resultado relativamente benigno ocurren más comúnmente en mujeres y aquellos con proteinuria en el rango no nefrótico. El tratamiento es controversial.
Glomeruloesclerosis segmentaria focal
La glomerulonefritis segmentaria focal se caracteriza por esclerosis (es decir, depósito incrementado de colágeno) de algunos, pero no todos los glomérulos y en el glomérulo afectado, y sólo se ve afectada una porción del ovillo glomerular. Es una causa particularmente común de síndrome nefrótico en hispanos y afroamericanos.
Aunque las esclerosis segmentaria focal es a menudo un síndrome idiopático, podría relacionarse con disminución de oxígeno en la sangre (ej. enfermedad de células falciformes ycardiopatía congénita cianótica), infección por virus de inmunodeficiencia humana (VIH) o abuso de drogas intravenosas, o podría ocurrir como un suceso secundario que refleja cicatrización glomerular debido a otras formas de glomerulonefritis.
La presencia de hipertensión y disminución de la función renal distingue a la esclerosis focal de la enfermedad de cambios mínimos. Además, la investigación indica que la excreción urinaria de CD80 (B7.1) es elevada con la enfermedad de cambios mínimos, pero no con la glomerulonefritis segmentaria focal. El trastorno se trata normalmente con corticosteroides. La mayoría de las personas con el trastorno progresan a insuficiencia renal en 5 a 10 años.
Hematuria o proteinuria asintomática
Muchos casos de glomerulonefritis producen enfermedad asintomática leve que no es reconocida o presentada a un profesional del cuidado de la salud y, por lo tanto, permanece sin diagnóstico. Los estudios de detección basados en la población han mostrado que el daño renal evidenciado por proteinuria, hematuria, TFG baja o una combinación de estas características está presente en la población. Los trastornos, tales como el púrpura de Henoch-Schönlein, desaparecen a menudo sin daño renal permanente, mientras que otros, tales como la nefropatía por IgA y el síndrome de Alport, pueden avanzar a enfermedad renal crónica e insu ciencia renal.
Nefropatía por inmunoglobulina A
La nefropatía por inmunoglobulina A (es decir, enfermedad de Berger) es una glomerulonefritis primaria caracterizada por la presencia de depósitos glomerulares de complejos inmunes de IgA. Puede ocurrir a cualquier edad, pero más comúnmente, la edad pico de diagnóstico es entre los 15 y 30 años de edad. La enfermedad ocurre con mayor frecuencia en varones que en mujeres y es la causa más común de nefritis glomerular en asiáticos.
El trastorno se caracteriza por depósito de complejos inmunes que contienen IgA en el mesangio del glomérulo. Una vez depositados en el riñón, los complejos inmunes se relacionan con inflamación glomerular. La causa del trastorno se desconoce y hay necesidad de clasificaciones más específicas de las etapas de nefropatía por IgA para poder interpretar más información. Por lo tanto, la Red Internacional de Nefropatía por IgA está desarrollando clasificaciones IgAN para ayudar a los proveedores a diagnosticar esta enfermedad. Algunas personas con el trastorno tienen concentraciones séricas altas de IgA.
Al principio de la enfermedad, muchas personas con el trastorno no tienen síntomas obvios y no están conscientes del problema. En estas personas, la nefropatía por IgA se sospecha durante la detección o examen rutinario de otra afección. En otras personas, el trastorno se presenta con hematuria macroscópica que va precedida de infección de vía respiratoria superior, síntomas del tubo digestivo o enfermedad parecida a la influenza. La hematuria dura por lo general 2 a 6 días.
Aproximadamente la mitad de las personas con hematuria macroscópica tienen un solo episodio, mientras que el resto experimenta un avance gradual de la enfermedad con episodios recurrentes de hematuria y proteinuria leve. El avance es por lo general lento, extendiéndose durante varias décadas.
La microscopia de inmunofluorescencia es esencial para el diagnóstico de nefropatía por IgA. El hallazgo diagnóstico es tinción mesangial para IgA más intensa que la tinción para IgG o IgM (figura 41-15). En el presente, no hay medias de tratamiento satisfactorias para la nefropatía por IgA.
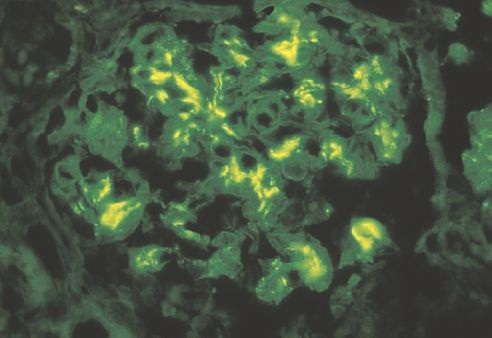
El rol de los fármacos inmunodepresivos tales como los esteroides y fármacos citotóxicos no está claro. Ha habido interés reciente en el empleo de ácidos grasos omega 3 para retardar el avance de la enfermedad.
Nefritis púrpura de Henoch-Schönlein
El púrpura de Henoch-Schönlein es una vasculitis de vasos pequeños que causa un exantema purpúrico en gran medida de las extremidades inferiores, artritis o artralgia, dolor abdominal y afectación renal idéntica a la de la nefropatía por IgA. La enfermedad se ve muy comúnmente en niños pero puede ocurrir también en adultos. La afectación renal no siempre está presenta al inicio, pero su incidencia se incrementa con el tiempo y es más común en niños mayores, quienes tienen dolor abdominal relacionado y exantema persistente. Aunque la hematuria y proteinuria son la presentación más común, algunas personas presentan manifestaciones de nefritis aguda y otras podrían presentar nefritis combinada con manifestaciones nefróticas. La mayoría de las personas se recuperan por completo durante un período de varias semanas. Los corticosteroides son el tratamiento más efectivo y se ha hallado que disminuyen la duración e intensidad del dolor abdominal y de articulaciones.
Síndrome de Alport
El síndrome de Alport representa un efecto hereditario de la MBG que da como resultado hematuria y podría avanzar a insuficiencia renal crónica. Tiende a relacionarse con defectos en los oídos u ojos. El síndrome es causado por mutaciones de colágeno tipo IV. Aproximadamente 85% de los casos son heredados como un rasgo dominante autosómico ligada al X, mientras que otros tienen patrones dominantes y recesivos autosómicos de la herencia. En las genealogías ligadas al X, la afectación es normalmente más grave en los niños que en las niñas. Los niños afectados avanzan por lo general a insuficiencia renal como los adultos. pero el avance podría ocurrir durante la adolescencia. Aunque muchas niñas nunca tienen más que hematuria leve con o sin proteinuria leve, algunas tienen enfermedad más significativa y podrían avanzar a insuficiencia renal.
El diagnóstico del síndrome de Alport se hace con frecuencia después del examen de la orina de un niño de una familia con casos múltiples de nefritis hereditaria. Los niños podrían presentar inicialmente hematuria microscópica intensa, seguida del desarrollo de proteinuria. Muchas, pero no todas, personas con síndrome de Alport tienen sordera sensorineural y varios trastornos oculares, incluso dislocación del cristalino, cataratas posteriores y distrofia corneal. La pérdida del oído es bilateral y con frecuencia se detecta primero durante la adolescencia.
Glomerulonefritis crónica
La glomerulonefritis crónica representa la fase crónica de varios tipos específicos de glomerulonefritis. Algunas formas de glomerulonefritis aguda (ej. glomerulonefritis postestreptocócica) experimentan resolución completa, mientras que otras avanzan a tasas variables de glomerulonefritis crónica. Algunas personas que presentan glomerulonefritis crónica no tienen historial de enfermedad glomerular. Estos casos podrían representar el resultado final de formas relativamente asintomáticas de glomerulonefritis. Histológicamente, la afección se caracteriza por riñones pequeños con glomérulos esclerosados. En muchos casos, la glomerulonefritis crónica se desarrolla de modo insidioso y avanza lentamente a enfermedad renal crónica en un período de años.
Lesiones glomerulares relacionadas con enfermedad sistémica
Muchos trastornos inmunitarios, metabólicos o hereditarios sistémicos se relacionan con lesión glomerular. En algunas enfermedades, tales como LES, diabetes mellitus e hipertensión, la afectación glomerular podría ser una manifestación clínica mayor.
Glomerulonefritis por lupus eritematoso sistémico
La afectación renal es clínicamente evidente en un 40% a un 85% de personas con LES y se observa más comúnmente en mujeres negras. La patogénesis del LES es incierta, pero parece estar relacionada con inmunidad desregulada de células B con producción de autoanticuerpos para diversos componentes nucleares, citoplásmicos, de la matriz extracelular y de la membrana celular.
La mayor parte de la lesión glomerular es activada por la formación de complejos inmunes dentro de la pared capilar glomerular.
Manifestaciones clínicas
Las manifestaciones clínicas del lupus eritematoso dependen del sitio de la lesión mediada por complejos inmunes. Los complejos inmunes confinados al mesangio causan menos inflamación que los complejos inmunes subendoteliales, los cuales tienen mayor exposición a células inflamatorias y mediadores en la sangre y, por lo tanto, tienen más probabilidades de producir inflamación. La Organización Mundial de la Salud (OMS) clasifica las lesiones glomerulares renales del LES como clase I, normal; clase II, proliferación mesangial; clase III, proliferación focal y segmentaria; clase IV, proliferación difusa; y clase I, proliferación membranosa.
Diagnóstico y tratamiento
Debido al alto riesgo de enfermedad renal, las personas con LES debe experimentar el análisis de orina rutinario para monitorear la apariencia de hematuria o proteinuria. Si se observan anomalías, se realiza a menudo biopsia renal. El tratamiento depende del grado de afectación glomerular. Las personas con glomerulonefritis clase I o II, por lo general, no requieren tratamiento. El avance a clases superiores va acompañado normalmente por un incremento en la actividad serológica del lupus y evidencia del deterioro de la función renal (es decir, aumento de creatinina sérica y una disminución de la TFG calculada). Los corticosteroides orales y los inhibidores de la enzima convertidora de angiotensina (ECA) son los pilares del tratamiento. Las personas con enfermedad más avanzada podrían requerir tratamiento con fármacos inmunodepresores (ej. ciclofosfamida intravenosa o mofetil micofenolato oral). Los estudios clínicos con otros fármacos inmunosupresores están en curso.
Glomeruloesclerosis diabética
La nefropatía diabética es una causa principal de enfermedad renal crónica y la causa más común de insuficiencia renal tratada mediante tratamiento de reemplazo renal en Estados Unidos. Ocurre enambos tipos 1 y 2 de diabetes mellitus. Es más prevalente entre afroamericanos, asiáticos y americanos nativos que entre blancos.
Fisiopatología
Las lesiones de nefropatía diabética afectan con mucha frecuencia a los glomérulos.
El engrosamiento extendido de la membrana basal capilar glomerular ocurre en casi todas las personas con diabetes y puede ocurrir sin evidencia de proteinuria. Esto va seguido de un incremento difuso de la matriz mesangial, con proliferación leve de células mesangiales. A medida que avanza la enfermedad, las células mesangiales chocan con el lumen capilar, reduciendo al área superficial para filtración glomerular. En la glomeruloesclerosis nodular, conocida también como síndrome de Kimmelstiel-Wilson, hay depósito nodular de hialina en la porción mesangial del glomérulo. A medida que el proceso esclerótico avanza en las formas difusa y nodular de la glomeruloesclerosis, hay obliteración completa del glomérulo, con deterioro de la función renal.
Aunque los mecanismos del cambio glomerular en la diabetes son inciertos, se cree que representan la síntesis mejorada o defectuosa de la MBG y la matriz mesangial con incorporación inapropiada de glucosa en los componentes no celulares de estas estructuras glomerulares.
Alternativamente, los cambios hemodinámicos que ocurren secundarios a los niveles altos de glucosa sanguínea podrían contribuir al inicio y avance de la glomeruloesclerosis diabética. Se ha establecido la hipótesis de que los aumentos de glucosa sanguínea producen un incremento en la TFG y la presión glomerular que conduce a un agrandamiento de los poros capilares glomerulares mediante un mecanismo que es, por lo menos en parte, mediado por angiotensina II. Este agrandamiento da como resultado un incremento en el contenido proteínico del filtrado glomerular, el cual a su vez requiere endocitosis incrementada de las proteínas filtradas mediante las células endoteliales tubulares, un proceso que en última instancia da lugar a destrucción de nefronas y deterioro progresivo de la función renal.
Manifestaciones clínicas y tratamiento
Las manifestaciones clínicas de la glomeruloesclerosis diabética se relacionan estrechamente con las de la diabetes. La TFG incrementada ocurre en personas con alteraciones tempranas en la función renal se relaciona con microalbuminuria, que se define como una excreción de albúmina urinaria de 30 mg a 300 mg en 24 h. La microalbuminuria es un factor predisponente importante de nefropatías diabéticas futuras. En muchos casos, los primeros cambios de la función glomerular pueden revertirse mediante el control cuidadoso de los niveles de glucosa sanguínea. Se ha demostrado que la inhibición de angiotensina por los inhibidores de la ECA o bloqueadores de receptores de angiotensina (BRA) tiene un efecto benéfico, posiblemente revirtiendo la presión glomerular incrementada. La hipertensión y el tabaquismo ha sido relacionados con el avance de la nefropatía diabética.
Enfermedad glomerular hipertensiva
La hipertensión leve a moderada causa cambios escleróticos en las arteriolas renales y arterias pequeñas, denominada nefroesclerosis benigna. Es muy frecuente y agresiva entre negros. Entre los afroamericanos, la hipertensión es la causa principal de enfermedad renal de etapa terminal.
La nefropatía hipertensiva se relaciona con varios cambios en la estructura y función renal. Los riñones son más pequeños de lo normal y son afectados por lo general bilateralmente. En el examen histológico, hay estrechamiento de las arteriolas y arterias pequeñas, causado por el engrosamiento y cicatrización de las paredes vasculares. A medida que se engrosan las estructuras vasculares y disminuye la perfusión, el flujo sanguíneo hacia la nefrona disminuye, causando atrofia tubular irregular, fibrosis intersticial y diversos cambios en la estructura y función glomerular.
Aunque la nefroesclerosis hipertensiva no complicada no se relaciona normalmente con anomalías significativas de la función renal, pocas personas podrían avanzar a enfermedad renal de etapa terminal. En riesgo particular de desarrollo de insuficiencia renal están tres tipos de personas: afroamericanos, personas con elevaciones de la TS más graves y personas con una segunda enfermedad subyacente, tales como la diabetes.