01. Trastornos congénitos y heredados de los riñones
Trastornos congénitos de los riñones
El riñón comienza a desarrollarse al inicio en la quinta semana de gestación y comienza a funcionar en las semanas 9 a 12 de gestación. En la semana 32, la producción fetal de orina alcanza 28 ml/h, aproximadamente. La orina que se produce se excreta en la cavidad amniótica y es el constituyente principal del líquido amniótico. Así, la cantidad relativa de líquido amniótico puede proporcionar información acerca del estado de la función renal fetal. En embarazos que afectan a lactantes con riñones no funcionales u obstrucción de flujo de orina desde los riñones, la cantidad de líquido amniótico es pequeña. Esta afección se llama oligohidramnios. Causa compresión del feto en desarrollo y se relaciona con frecuencia con el desarrollo afectado de los pulmones y otras estructuras fetales.
Las anomalías de forma y posición son los problemas renales congénitos más comunes. Menos comunes son los trastornos relacionados con una disminución de la masa renal (ej. agénesis e hipogénesis) o un cambio de la estructura renal (ej. displasia renal). Los riñones pueden verse tan pronto como a las 12 semanas de gestación mediante ultrasonografía, permitiendo que muchas anomalías urinarias fetales se detecten antes del nacimiento.
Agénesis e hipoplasia
El término disgénesis se refiere al fallo de un órgano para desarrollarse normalmente. Agénesis se refiere al fallo de un órgano para desarrollarse en absoluto. La agénesis renal lateral es relativamente común. Ocurre en cerca de 1 de 1.000 a 2.000 lactantes recién nacidos. Los niños son afectados con mayor frecuencia que las niñas. La agénesis unilateral normalmente no causa síntomas En muchos casos no se descubre durante la infancia porque el otro riñón experimenta normalmente hipertrofia compensatoria y realiza la función del riñón faltante.
La agénesis total de ambos riñones es incompatible con la vida extrauterina. Los lactantes son mortinatos o mueren poco después del nacimiento por hipoplasia pulmonar. Los recién nacidos con agénesis renal, con frecuencia, tienen rasgos faciales característicos, algunas veces llamados síndrome de Potter, que resultan de los efectos de oligohidramios. Los ojos están ampliamente separados y tienen pliegues epicánticos, los oídos están abajo de su lugar, la nariz es ancha y plana, la barbilla es plana y los defectos de las extremidades con frecuencia están presentes.
En la hiperplasia renal, los riñones no se desarrollan al tamaño normal. Al igual que la agénesis, la hipoplasia afecta con más frecuencia sólo un riñón. Cuando ambos riñones son afectados, hay desarrollo progresivo de insuficiencia renal. Se ha sugerido que la hipoplasia verdadera es en extremo rara. La mayoría de los casos representan probablemente nefroesclerosis adquirida debido a enfermedades vasculares, infecciosas u otras enfermedades renales en vez de una insuficiencia del desarrollo subyacente.
Displasia renal
La displasia renal es causada por una anomalía en la diferenciación de las estructuras del riñón durante el desarrollo embrionario. Se caracteriza por estructuras tubulares no diferenciadas rodeadas por tejido embriónico primitivo. El trastorno podría dar como resultado riñones aplásticos pequeños o quistes que se forman de los túbulos anómalos. Si los quistes están presentes, la afección se denomina displasia quística. Uno o ambos riñones podrían estar afectados y el riñón afectado podría ser irregularmente grande o pequeño. Muchas formas de displasia van acompañadas de anomalías de las vías urinarias, en particular los trastornos que causan obstrucción al flujo de orina (ej. agénesia uretral o atresis, obstrucción de la unión ureteropélvica).
Un riñón multiquístico es aquél en el que el riñón se reemplaza por quistes y no funciona. El riñón no tiene la forma usual, sino que es, además, una masa de quistes. La displasia renal multiquística unilateral es la causa más común de una masa abdominal en recién nacidos. La función del riñón opuesto es, por lo común, normal y estos niños tienen un excelente pronóstico después de la remoción quirúrgica del riñón afectado. La displasia renal bilateral causa oligohidramnios y los rasgos faciales de Potter resultantes, hipoplasia pulmonar e insuficiencia renal.
Alteraciones en la posición y forma del riñón
El desarrollo de los riñones a lo largo de la vida embrionaria puede dar como resultado riñones ectópicos que yacen fuere de su posición normal. Uno o ambos riñones podrían estar en posición anómala. La mayoría de riñones ectópicos se localizan justo arriba del borde pélvico o dentro de la pelvis, pero algunos yacen en la parte inferior del abdomen. Debido a la posición anómala, podría ocurrir retorcimiento de los uréteres y obstrucción del flujo urinario.
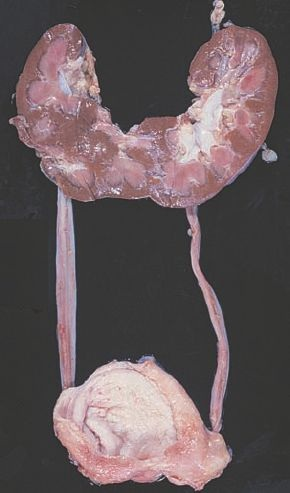
Una de las alteraciones más comunes en la forma del riñón es una anomalía llamada riñón en forma de herradura. Esta anomalía ocurre en, aproximadamente, 1 de cada 500 a 1.000 personas. En este trastorno, los polos superior e inferior de los riñones se fusionan, produciendo una estructura en forma de herradura que es continua a lo largo de la línea media del cuerpo anterior a los grandes vasos (figura 41-1). La afección normalmente no causa problemas, a menos que haya un defecto relacionado en la pelvis renal u otras estructuras urinarias que obstruya el flujo urinario.
Enfermedades renales quísticas heredadas
Las enfermedades renales quísticas heredadas, que son trastornos de un solo gen y son heredadas como rasgos mendelianos, incluyen la enfermedad renal dominante autosómica y poliquística recesiva, y la enfermedad quística de nefronoptisis-medular. Las enfermedades renales poliquísticas son un grupo de trastornos del riñón caracterizados por sacos llenos de líquido o segmentos que tienen su origen en las estructuras tubulares del riñón. Los quistes podrían ser uno solo o varios y pueden variar de tamaño desde microscópicos hasta varios centímetros de diámetro. Aunque podrían surgir como una anomalía del desarrollo o ser adquiridos después en la vida, la mayoría de las formas son hereditarias.
En la forma dominante autosómica de la enfermedad renal poliquística (ERPQ), miles de quistes grandes se derivan de cada segmento de la nefrona (figura 41-2). La pared tubular, que está revestida por una sola capa de células tubulares, expande y luego se cierra rápidamente el quiste desde el túbulo de origen. En la forma recesiva autosómica de la enfermedad renal poliquística (ERPQ), pequeños quistes alargados se forman en los conductos recolectores y mantienen contacto con la nefrona de origen (figura 41-2).
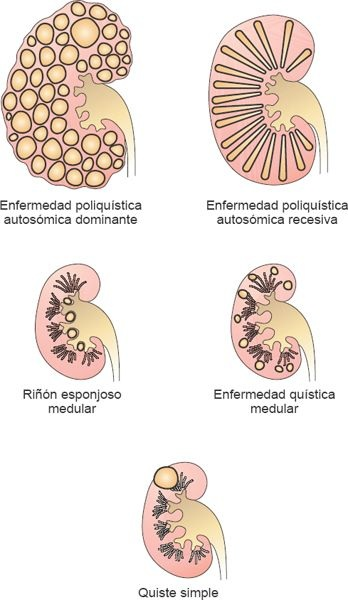
En la nefronoptisis, enfermedad renal quística medular, los quistes están restringidos al borde corticomedular. Los quistes simples son quistes adquiridos que se desarrollan en el riñón como una consecuencia del envejecimiento, diálisis u otras afecciones que afectan la función tubular.
Enfermedad renal poliquística dominante autosómica
La enfermedad renal poliquística dominante autosómica, conocida también como enfermedad poliquística del adulto, es la forma más común de enfermedad quística renal. El trastorno, que es heredado como un rasgo autosómico, da como resultado la formación de quistes destructivos llenos de líquido en el riñón y otros órganos. La ERPQ afecta a más de 1:400 a 1:1.000 personas en Estados Unidos. La mitad de estas personas desarrollan finalmente la enfermedad renal de etapa terminal (ERET). La enfermedad explica el 5% de todos los casos de enfermedad renal crónica que requieren diálisis o trasplante.
Hay 2 tipos de ERPQ:
- Tipo I, causada por mutaciones en el gen PKD1, y explica el 85% de los casos.
- Tipo II, causada por mutaciones en el gen PKD2, y explica la mayor parte del 15% restante de los casos.
Los productos de estos genes, policistina-1 y policistina-2, se encuentran en los cilios primarios que revisten la superficie apical del epitelio tubular. Se cree que estos cilios primarios actúan como sensores del flujo urinario y como transductores de señal para la proliferación celular tubular,diferenciación y apoptosis.
Causas y patogénesis
Aunque la patogénesis de la ERPQ no es clara, se cree que los quistes surgen en segmentos de los túbulos renales de unas cuantas células epiteliales. Las células epiteliales que revisten los quistes de la ERPQ tienen una alta tasa de proliferación y son relativamente no diferenciados. De modo concurrente, una membrana basal defectuosa inmediatamente subyacente al epitelio anómalo permite la dilatación y formación del quiste. Los quistes se desprenden frecuentemente de las células del revestimiento epitelial.
Históricamente, se pensaba que la enfermedad renal crónica resultaba de la presión ejercida por los quistes en expansión sobre el tejido renal circundante normal. Sin embargo, se reconoce ahora que los quistes surgen en menos de 2% de las nefronas y que factores distintos a la compresión de los quistes en expansión explican la pérdida de tejido renal funcional.
En la actualidad, se piensa que la pérdida apoptótica de las células tubulares renales y la acumulación de mediadores inflamatorios contribuyen a la destrucción del tejido renal normal. Adicionalmente, los factores de crecimiento angiogénicos se relacionan con la gravedad de la enfermedad, en particular en personas jóvenes que tienen ERPQ. Las mutaciones en los genes PKD1 y PKD2 producen enfermedad renal y extrarrenal idéntica, pero el avance de la enfermedad es, por lo general, más rápido en personas con enfermedad tipo I.
Manifestaciones clínicas
Normalmente, el avance de la enfermedad renal es lento y la ERET es poco común para adultos antes de los 40 años de edad. Inicialmente, los quistes son, por lo general, asintomáticos y la función (renal y) hepática es normal.
Conforme avanza la enfermedad del riñón, las manifestaciones de la ERPQ incluyen dolor por agrandamiento de los quistes que podrían alcanzar niveles debilitantes, episodios de hematuria macroscópica de hemorragia hacia un quiste, quistes infectados de ITU ascendente e hipertensión que resulta de la compresión de vasos sanguíneos intrarrenales con activación del mecanismo renina-angiotensina.
Por lo general, los riñones están agrandados en personas con ERPQ y pueden alcanzar tamaños enormes (figura 41-3). Los contornos externos de los riñones son distorsionados por numerosos quistes, algunos tan grandes como 5 cm de diámetro, que están llenos de líquido color paja. Los quistes podrían hallarse en el hígado y, con menor frecuencia, en el páncreas y el bazo.
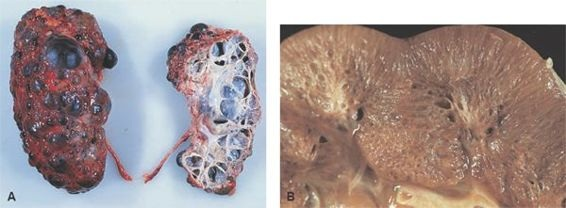
A medida que la enfermedad continúa avanzando, las manifestaciones extrarrenales tales como aneurismas son frecuentes, enfatizando la naturaleza sistémica de la enfermedad. Aproximadamente 20% de las personas con enfermedad renal poliquística tienen un aneurisma relacionado, y la hemorragia subaracnoidea es una causa frecuente de muerte.
Diagnóstico y tratamiento
No se ha hallado aún que los niveles de creatinina sérica sean un marcador efectivo del empeoramiento de la ERPQ, pero se ha determinado que la excreción de albúmina en la orina (EAO) es un factor predisponente confiable, así como el incremento de electrolitos y la hematuria.La ultrasonografía es el técnica preferida para diagnóstico de ERPQ en personas sintomáticas y para detección de miembros de la familia asintomáticos. La tomografía computarizada (TC) podría utilizarse para la detección de quistes pequeños. Los estudios de ligamiento genético se utilizan para el diagnóstico de ERPQ, pero, por lo general, se reservan para casos en los que la imagen radiográfica es negativa y es esencial la necesidad de un diagnóstico definitivo, tal como cuando se selecciona a los miembros de la familia para posible donación de riñón.
El tratamiento de la ERPQ es, en gran medida, de apoyo y va dirigido a retardar el avance de la enfermedad. El fármaco, Tolvaptan, que es un antagonista selectivo del receptor de vasopresina V2, ha sido estudiado y se halló que inhibe el desarrollo quístico y conserva la función del riñón. El control de la hipertensión y prevención de las IVU son importantes. El dolor es una queja común de las personas con ERPQ. Por lo tanto, se necesita un enfoque sistémico para diferenciar la causa del dolor y Definir un método para el control. La diálisis y trasplante de riñón se reservan para quienes avanzan hacia la insuficiencia renal. Sin embargo, es importante observar que la diálisis prolongada incrementará la formación de quistes incluso en personas con ERPQ.
Enfermedad renal poliquística recesiva autosómica
La enfermedad renal poliquística recesiva autosómica se caracteriza por dilatación quística de los túbulos recolectores corticales y medulares(figura 41-2). Es rara en comparación con la ERPQ. La ERPQ es causada por mutaciones en el gen PKHD1. El producto del gen, fibrocistina, se encuentra en el riñón, hígado y páncreas, y al parecer participa en la regulación de la proliferación y adhesión celular.
Manifestaciones clínicas
El lactante típico con ERPQ se presenta con masas bilaterales en el flanco, acompañado por insuficiencia renal grave, signos de desarrollo pulmonar deteriorado y grados variables de fibrosis hepática e hipertensión portal. Las facies de Potter y otros defectos relacionados con oligohidramnios podrían estar presentes. La hipertensión se observa normalmente dentro de las primeras semanas de vida y, con frecuencia, es grave. Muchos lactantes mueren durante el período perinatal, a menudo de hipoplasia pulmonar. Los casos excepcionales de ERPQ se manifiestan en niños y adultos.
Tratamiento
El tratamiento de la ERPQ es, en gran medida, de apoyo. El soporte ventilatorio dinámico es, a menudo, necesario en el período neonatal debido a la hipoplasia e hipoventilación pulmonar. Las modernas técnicas respiratorias neonatales y el tratamiento de reemplazo renal han incrementado la tasa de supervivencia de 10 años que sobreviven más allá del primer año de vida. La morbididad y mortalidad en niños mayores se relaciona con complicaciones de insuficiencia renal crónica y enfermedad hepática.
Complejo nefronoptisis-enfermedad quística medular
El complejo de nefronoptisis-enfermedad quística medular es un grupo de trastornos renales que tienen su inicio en la infancia. Las características comunes son riñones pequeños y encogidos y la presencia de un número variable de quistes concentrados, por lo general, en la unión corticomedular. El ataque inicial es en los túbulos distales, con perturbación de la membrana basal tubular seguida de atrofia tubular crónica y progresiva que afecta la médula y la corteza. Aunque la presencia de quistes medulares es importante, el daño cortical y tubular es la causa eventual de enfermedad e insuficiencia renal crónica.
Como un complejo, los trastornos explican, del 19% al 25% de la insuficiencia renal en la infancia. Los niños afectados presentan primero poliuria, polidipsia y enuresis (incontinencia nocturna), lo cual refleja la capacidad afectada de los riñones para concentrar orina. Otras manifestaciones de los trastornos incluyen pérdida de sal, retardo del crecimiento, anemia e insuficiencia renal progresiva. Algunas formas juveniles de nefronoptisis tienen complicaciones extrarrenales, incluso anomalías motoras oculares, retinitis pigmentosa, fibrosis hepática y anomalías cerebelares. La azoemia progresiva y la insuficiencia renal siguen, por lo general, en 5 a 10 años.
Quistes renales simples y adquiridos
Los quistes simples son un trastorno común del riñón. Éstos podrían ser simples o múltiples, unilaterales o bilaterales y, por lo general, son menores que 1 cm de diámetro, aunque podrían crecer más grandes. La mayoría de quistes simples no producen signos o síntomas, o comprometen la función renal. Cuando son asintomáticos, los quistes podrían causar dolor lumbar, hematuria, infección e hipertensión relacionados con la estimulación producida por isquemia del sistema renina-angiotensina. Son los más comunes en adultos mayores. Aunque los quistes son benignos, podrían confundirse con carcinoma celular renal.
Una forma adquirida de enfermedad quística renal ocurre en personas con insuficiencia renal de etapa terminal (IRET) que han experimentado tratamiento prolongado con diálisis. Aunque la afección es en gran medida asintomática, los quistes podrían sangrar, causando hematuria. Los tumores, por lo general adenomas pero ocasionalmente adenosarcomas, podrían desarrollarse en las paredes de estos quistes.