Trastornos de la hemostasia
Hemostasia se refiere a la detención del flujo sanguíneo. El proceso normal de la hemostasia se regula mediante una disposición compleja de activadores e inhibidores que mantienen la fluidez sanguínea y evitan que la sangre salga del compartimiento vascular. La hemostasia es normal cuando sella un vaso sanguíneo para evitar la pérdida de sangre y la hemorragia. Es anómala cuando causacoagulación sanguínea inapropiada o cuando la coagulación es insuficiente para detener el flujo de sangre desde el compartimiento vascular. Los trastornos de la hemostasia caen en 2 categorías principales: formación inapropiada de coágulos dentro del sistema vascular (trombosis) e incapacidad de la sangre para coagularse en respuesta a un estímulo apropiado (hemorragia).
Conceptos clave
Los conceptos clave proporcionan un breve resumen de los aspectos más importantes de cada uno de los apartados correspondientes dentro del capítulo. Si alguno de estos puntos no está claro, acuda al apartado correspondiente para su revisión.
Hemostasia es el proceso ordenado por pasos para detener la hemorragia que incluye vasoespasmo, formación de un tapón plaquetario y desarrollo de un coágulo de fibrina.
El proceso de coagulación sanguínea requiere la presencia de plaquetas producidas en la médula ósea, FvW generado por el endotelio vascular y factores de coagulación sintetizados en el hígado, con la utilización de vitamina K.
Los trombos arteriales se relacionan con padecimientos que producen flujo sanguíneo turbulento y adherencia plaquetaria.
Los trombos venosos se vinculan con afecciones que causan estasis del flujo sanguíneo con concentraciones incrementadas de factores de coagulación.
Los trastornos de formación del tapón plaquetario incluyen una disminución en la cantidad de plaquetas debida a su producción inadecuada de éstas (disfunción de médula ósea), exceso de destrucción de plaquetas (trombocitopenia), función plaquetaria anómala (trombocitopatía) o defectos en el FvW.
La alteración de la etapa de coagulación de la hemostasia se debe a una insuficiencia de uno o más de los factores de coagulación.
Los trastornos de la integridad de los vasos sanguíneos son resultado de vasos estructuralmente débiles o lesión del vaso por inflamación y mecanismos inmunitarios.
Resumen
Este apartado es un resumen del capítulo que le ayudará a repasar el núcleo fundamental de la materia de cara a un hipotético examen.
Mecanismos de la hemostasia
La hemostasia está diseñada para mantener la integridad del compartimiento vascular.
El proceso se divide en 3 fases:
- vasoconstricción del vaso, que restringe el tamaño del vaso y reduce el flujo de sangre;
- adhesión plaquetaria y formación del tapón de plaquetas; y
- formación del coágulo de fibrina, que une el tapón de plaquetas.
La retracción del coágulo, que tira de los bordes del vaso lesionado y, su disolución, que implica la acción de la plasmina para desintegrarlo y permitir que el flujo de sangre se restablezca y que la cicatrización tisular tenga lugar, también son procesos importantes de la hemostasia.
La coagulación sanguínea requiere la activación por pasos de los factores de coagulación, que son controlados con cuidado por activadores e inhibidores.
Estados de hipercoagulabilidad
La hipercoagulabilidad causa coagulación excesiva y contribuye a la formación de trombos. Es resultado de afecciones que promueven un incremento del número o la función de las plaquetas, o actividad acelerada del sistema de coagulación. La trombocitosis, una elevación del recuento de plaquetario, puede presentarse como un proceso reactivo (trombocitosis secundaria) o un proceso esencial (trombocitosis primaria).
El aumento de la función plaquetaria suele deberse a trastornos, como la ateroesclerosis, que dañan el endotelio vascular y perturban el flujo sanguíneo, o a condiciones que incrementan la sensibilidad de las plaquetas a factores que promueven la adhesividad y agregación, como el tabaquismo.
Los factores que aceleran la actividad del sistema de coagulación incluyen estasis del flujo sanguíneo, cuyo resultado es una acumulación de factores de coagulación y alteraciones en los componentes del sistema de coagulación (es decir, incremento de los factores procoagulantes o disminución de los factores anticoagulantes). El síndrome antifosfolípido, un trastorno de coagulación venosa y arterial adquirido, se manifiesta como trastorno primario o como trastorno secundario vinculado con lupus eritematoso sistémico. Se relaciona con anticuerpos antifosfolípidos, que promueven la trombosis que puede afectar muchos órganos.
Trastornos hemorrágicos
Los trastornos hemorrágicos o el deterioro de la coagulación sanguínea pueden ser consecuencia de defectos en cualquiera de los factores que contribuyen a la hemostasia: plaquetas, factores de coagulación o integridad vascular.
El número de plaquetas circulantes puede reducirse (es decir, trombocitopenia) por producción reducida de la médula ósea, acumulación excesiva en el bazo o destrucción inmunitaria.
La función plaquetaria defectuosa (es decir, trombocitopatía) es causada por trastornos heredados (enfermedad de von Willebrand) o por medicamentos o enfermedad.
Es posible que el deterioro de la coagulación sanguínea se deba a las insuficiencias de uno o más factores de coagulación conocidos.
Las insuficiencias pueden ser secundarias a trastornos adquiridos (es decir, enfermedad hepática odeficiencia de vitamina K) o enfermedades heredadas (es decir, hemofilia A o enfermedad de von Willebrand).
La hemorragia también tiene lugar en vasos estructuralmente débiles por alteraciones de la síntesis de componentes de la pared vascular (es decir, insuficiencia de vitamina C, niveles excesivos de cortisol como en la enfermedad de Cushing o el proceso de envejecimiento) o por daño secundario a mecanismos genéticos (es decir, telangiectasia hemorrágica), o la presencia de microtrombos.
La CID se distingue por coagulación generalizada y hemorragia en el compartimiento vascular. Comienza con activación masiva de la cascada de coagulación y generación de microtrombos que causan oclusión vascular e isquemia tisular. La formación del coágulo consume todas las proteínas y plaquetas de coagulación disponibles, y produce hemorragia grave.
Mecanismos de la hemostasia
La hemostasia se divide en 3 etapas:
- Constricción vascular.
- Formación del tapón plaquetario.
- Coagulación sanguínea (figura 26-1).
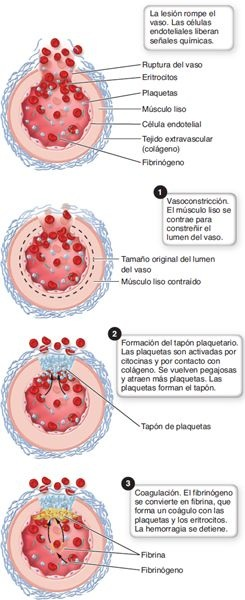
Durante el proceso de hemostasia, las hebras de fibrina similares al pelo pegan las plaquetas agregadas para formar la base estructural del coágulo sanguíneo. En presencia de fibrina, el plasma se vuelve gelatinoso y atrapa eritrocitos y otros elementos formados en la sangre. La hemostasia se completa cuando el tejido fibroso crece hacia el coágulo y sella el agujero en el vaso.
Constricción vascular
El espasmo de los vasos constriñe el vaso y reduce el flujo sanguíneo. Es un fenómeno transitorio que suele durar minutos u horas. El espasmo de los vasos se inicia por lesión endotelial y es causado por mecanismos locales y humorales. Los reflejos neurales y el tromboxano A (TXA de las plaquetas y otros mediadores, como la serotonina, contri-2), una prostaglandina liberada de las plaquetas y otros mediadores, como la serotonina, contribuyen a la vasoconstricción. El vasoconstrictor más poderoso es la endotelina. La prostaciclina, otra prostaglandina liberada del endotelio del vaso, produce vasodilatación e inhibe la agregación plaquetaria en el endotelio no lesionado circundante.
Formación del tapón plaquetario
El tapón plaquetario, la segunda línea de defensa, se inicia cuando las plaquetas entran en contacto con la pared vascular. Pequeñas rupturas en la pared del vaso a menudo se sellan con el tapón plaquetario y no con un coágulo de sangre.
Las plaquetas, o trombocitos, son grandes fragmentos del citoplasma de células de la médulaósea llamados megacariocitos.
La plaqueta tiene una vida media cercana a 8 a 12 días y luego los macrófagos se encargan de descomponerla y eliminarla. La concentración sérica normal se aproxima a 150.000 a 400.000 plaquetas por microlitro (μl) de sangre. La producción de plaquetas está controlada por una proteína, llamada trombopoyetina, que causa proliferación y maduración de megacariocitos. Las fuentes de trombopoyetina incluyen hígado, riñones, músculo liso y médula ósea.
Las plaquetas, poseen una membrana celular pero no núcleo y no pueden reproducirse. La membrana celular tiene fosfolípidos que ayudan en el proceso de coagulación. Aunque carecen de un núcleo, tienen muchas de las características de una célula completa. La membrana celular externa está cubierta por una capa de glucoproteínas, glucosaminoglucanos y proteínas de coagulación (figura 26-2).
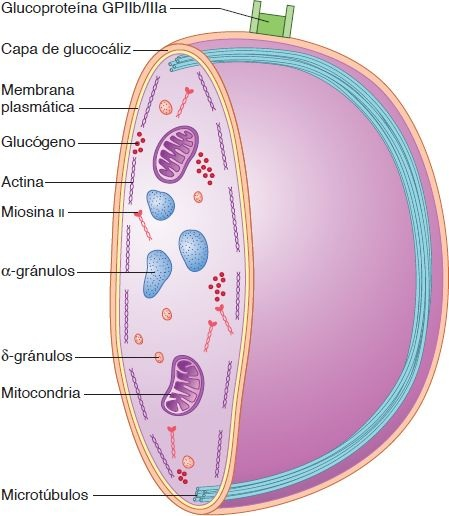
Una de las glucoproteínas importantes es GPIIb/IIIa, que se une al fibrinógeno y une las plaquetas entre sí. Microtúbulos y filamentos de actina y miosina que soportan la membrana celular mantienen la forma de la plaqueta. Las plaquetas tienen mitocondrias y sistemas enzimáticos capaces de producir trifosfato de adenosina (ATP) y difosfato de adenosina (ADP). Poseen también las enzimas necesarias para la síntesis de la prostaglandina, TXA2, que se requiere para su función en la hemostasia.
Las plaquetas tienen 2 tipos específicos de gránulos (α y θ) que liberan mediadores para la hemostasia. Los α-gránulos expresan selectina P, una proteína adhesiva en su superficie y contienen fibrinógeno, factor de von Willebrand (FvW), fibronectina, factores V y VIII, factor plaquetario (una quimiocina que se une a heparina), factor de crecimiento derivado de plaquetas (FCDP), factor de crecimiento transformador (FCT-α) y trombospondina. La liberación de factores de crecimiento da como resultado la proliferación y crecimiento de células endoteliales vasculares, células de músculo liso y fibroblastos, y es importante en la reparación vascular. Los θ-gránulos, o gránulos densos, contienen ADP y ATP, calcio ionizado, histamina, serotonina y adrenalina, los cuales contribuyen a la vasoconstricción.
La formación del tapón plaquetario implica adhesión y agregación de plaquetas. Las plaquetas son atraídas a la pared del vaso dañado, se activan y cambian de discos lisos a esferas espinosas, con lo que exponen los receptores de glucoproteína en sus superficies. La adhesión de plaquetas requiere una molécula de proteína llamada factor de von Willebrand, que se fuga hacia el tejido lesionado desde el plasma. Este factor es producido por las células endoteliales de los vasos sanguíneos y circula en la sangre como una proteína portadora para el factor de coagulación VIII. La adhesión a la capa subendotelial del vaso tiene lugar cuando el receptor de plaquetas se une al FvW en el sitio de la lesión, lo que liga la plaqueta a las fibras de colágeno expuestas.
La agregación plaquetaria ocurre poco después de la adhesión. La secreción del contenido de los gránulos de plaquetas actúa como mediadora. La liberación del contenido corporal denso tiene particular importancia porque se requiere calcio para el componente de coagulación de la hemostasia y el ADP es un mediador de la agregación plaquetaria. La liberación de ADP facilita también su liberación de otras plaquetas, lo cual conduce a la amplificación del proceso de agregación. Además de ADP, las plaquetas secretan la prostaglandina TXA2, que es un estímulo importante para la agregación plaquetaria. Las acciones combinadas de ADP y TXA2 conducen a la expansión del agregado plaquetario que se convierte en el tapón hemostático primario. El tapón plaquetario se estabiliza conforme la vía de coagulación se activa en la superficie plaquetaria y el fibrinógeno se convierte en fibrina. Esto crea una red de fibrina que cimienta las plaquetas y otros componentes sanguíneos juntos (figura 26-1). La selectina P también es parte del proceso de agregación plaquetaria puesto que se une a leucocitos, que, con sustancias plaquetarias como FCDP, participan en la cicatrización de la pared del vaso.
La membrana plaquetaria desempeña una función relevante en la adhesión de plaquetas y el proceso de coagulación. La cubierta de glucoproteínas en su superficie controla las interacciones con el endotelio vascular. En condiciones normales, las plaquetas evitan la adhesión al endotelio, pero interactúan con áreas lesionadas de la pared vascular y el colágeno expuesto más profundo. Los receptores de glucoproteína (GPIIb/IIIa) en la membrana plaquetaria se unen a fibrinógeno y vinculan plaquetas. La formación defectuosa del tapón plaquetario causa hemorragia en personas con insuficiencia de plaquetas o FvW. Además de sellar las rupturas vasculares, las plaquetas desempeñan una función casi continua en el mantenimiento de la integridad vascular normal. Pueden aportarfactores de crecimiento para las células musculares lisas endoteliales y arteriales. Las personas con insuficiencia plaquetaria tienen permeabilidad capilar incrementada y sufren pequeñas hemorragias cutáneas por traumatismos o cambios en la presión arterial.
Los inhibidores de la agregación plaquetaria, incluidos ácido acetilsalicílico, clopidogrel y ticlopidina, pueden consumirse para evitar la agregación plaquetaria y la formación de coágulos en personas que están en riesgo de infarto de miocardio, accidente cerebrovascular o enfermedad arterial periférica. El tratamiento con ácido acetilsalicílico en dosis bajas inhibe la síntesis de prostaglandina, incluida TXA2.
El clopidogrel y la ticlopidina alcanzan sus efectos plaquetarios al inhibir la vía de ADP en las plaquetas. A diferencia del ácido acetilsalicílico, estos fármacos tienen un efecto en la síntesis de prostaglandina. Tanto el clopidogrel como la ticlopidina prolongan el tiempo de hemorragia. Sin embargo, los efectos secundarios más graves de la ticlopidina son neutropenia y púrpura trombocitopénica trombótica.
Estudios de ensayos aleatorizados demostraron que el clopidogrel en combinación con ácido acetilsalicílico se relaciona con una reducción de accidentes cardíacos mayores, similar a combinar ticlopidina con ácido acetilsalicílico, y parecieron más seguros. Las Antiplatelet Therapy Guidelines publicadas de manera conjunta por la American Heart Association y el American College of Cardiology se actualizaron para sugerir que el empleo del inhibidor de la bomba de protones con tratamiento antiplaquetario, en específico con clopidogrel, podría causar efectos cardíacos adversos por la interacción del fármaco con el citocromo P450. Así, aunque agregar un inhibidor de la bomba de protones a la mayor parte del tratamiento antiplaquetario parece ser eficaz, se recomienda que el clopidogrel se administre sin un inhibidor de la bomba de protones por los posibles efectos secundarios cardiovasculares negativos.
Los fármacos que actúan como inhibidores del receptor GPIIb/IIIa (tirofibán, eptifibatida, abciximab) se desarrollaron para utilizarse en el tratamiento de síndromes coronarios agudos.
Coagulación sanguínea
La cascada de coagulación es parte del proceso hemostático. Es un proceso por pasos que promueve la conversión de la proteína plasmática soluble, el fibrinógeno, en fibrina. Las hebras de fibrina insoluble crean una red que pega las plaquetas y otros componentes de la sangre para formar el coágulo.
Muchas sustancias que promueven la formación del coágulo (factores de procoagulación) o lainhiben (factores de anti-coagulación) controlan el proceso de coagulación. Cada uno de los factores de procoagulación o coagulación, identificados con números romanos, realiza un paso específico en el proceso de coagulación. La activación de un factor de procoagulación o proenzima está diseñada para activar el siguiente factor en la secuencia (efecto de cascada). Como la mayoría de los factores de procoagulación inactivos está presente en la sangre todo el tiempo, el proceso de varios pasos asegura que no tenga lugar un episodio masivo de coagulación intravascular. Esto también significa que las anomalías del proceso de coagulación se presentan cuando uno o más de los factores son insuficientes o cuando las condiciones conducen a la activación inapropiada de alguno de los pasos.
Casi todos los factores de coagulación son proteínas sintetizadas en el hígado. La vitamina K es necesaria para la síntesis de factores II, VII, IX y X, protrombina y proteína C. Si hay insuficiencia de vitamina K o insuficiencia hepática que imposibiliten la creación de suficiente protrombina, se desarrolla una tendencia hemorrágica. El calcio (factor iv) es necesario en todos excepto los 2 primeros pasos del proceso de coagulación. El cuerpo suele tener cantidades suficientes de calcio para estas reacciones. La inactivación del ion calcio evita que la sangre se coagule cuando se elimina del cuerpo. La adición de citrato a la sangre almacenada para fines de transfusión evita la coagulación al formar un quelato con el ion calcio. El ácido etilendiaminotetraacético (AEDT), otro quelante, a menudo se agrega a muestras de sangre que se utilizan para análisis en el laboratorio clínico.
El proceso de coagulación es resultado de la activación de lo que tradicionalmente se designa como las vías intrínseca y extrínseca, las cuales forman el activador de protrombina (figura 26-3).
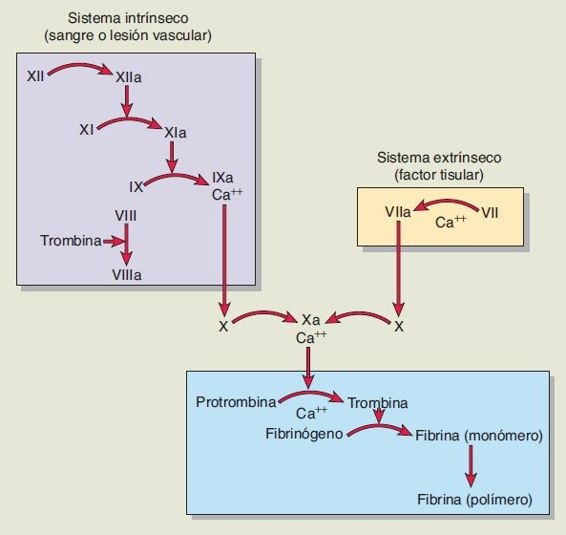
La vía intrínseca, que es un proceso hasta cierto punto lento (puede producir coagulación en 1 min a 6 min), comienza en la circulación con la activación del factor XII1.
La vía extrínseca, que es un proceso mucho más rápido (puede producir coagulación en 15 s), inicia con el traumatismo del vaso sanguíneo o los tejidos circundantes y la liberación del factor tisular o tromboplastina tisular, una lipoproteína adhesiva, de las células subendoteliales. Se compone de fosfolípidos de las membranas junto con un complejo de lipoproteína que actúa como una enzima proteolítica.
Los pasos terminales en ambas vías son los mismos: activación del factor X y conversión de protrombina en trombina. La trombina actúa luego como una enzima para convertir fibrinógeno en fibrina, el material que estabiliza un coágulo. Ambas vías son necesarias para la hemostasia normal y existen muchas interrelaciones entre ellas. Cada sistema se activa cuando la sangre sale del sistema vascular. El sistema intrínseco se activa cuando la sangre entra en contacto con el colágeno en la pared del vaso lesionado. El sistema extrínseco se activa cuando la sangre se expone a extractos tisulares. Sin embargo, la hemorragia secundaria por defectos en el sistema extrínseco no suele ser tan grave como la que resulta de defectos en la vía intrínseca.
La coagulación sanguínea está regulada por varios anti-coagulantes naturales. La antitrombina III inactiva los factores de coagulación y neutraliza la trombina, la última enzima en la vía para la conversión de fibrinógeno en fibrina. Cuando la anti-trombina III forma un complejo con la heparina de origen natural, su acción se acelera para inactivar la trombina, el factor Xa y otros factores de coagulación. Esta activación compleja confiere protección contra la formación de trombos no controlada en la superficie endotelial.
La proteína C, una proteína plasmática, actúa como un anti-coagulante al inactivar los factores V y VIII. El antígeno de proteína C, o antígeno PC (factor V Leiden), se produce en el hígado y previene la trombosis. La insuficiencia de proteína C es congénita del 35% al 58% de las veces, pero también puede ser adquirida si se tiene insuficiencia hepática grave, insuficiencia de vitamina K o neoplasia. Este trastorno es un defecto hereditario en el factor V y confiere mayor riesgo de coagulación. Puede medirse con una prueba de resistencia de proteína C y el intervalo normal debe estar entre 0,6 y 1,25 de antígeno PC normal. Las mujeres con factor V Leiden combinado con la influencia protrombótica del embarazo están en alto riesgo de tener resultados adversos de la gestación como trastornos de tromboembolia venosa, preeclampsia, pérdida del feto y desprendimiento de la placenta.
La proteína S, otra proteína plasmática, acelera la acción de la proteína C. Una insuficiencia de proteína C o S pone a los individuos en riesgo de trombosis. Se realiza una prueba de proteína S para determinar si la insuficiencia es heredada o adquirida porque a menudo las personas con trastornos autoinmunitarios están en riesgo de tener carencia de proteína S. El intervalo normal para las mujeres es de 0,5 a 1,2 de la actividad normal y para varones, de 0,6 a 1,3.
La plasmina descompone la fibrina en productos de degradación de fibrina que actúan como anticoagulantes. Se sugiere que algunos de estos anticoagulantes naturales podrían desempeñar una función en la hemorragia que se observa con coagulación intravascular diseminada (CID).
Los fármacos anticoagulantes warfarina y heparina se utilizan para prevenir trastornos tromboembólicos, como trombosis venosa profunda y embolismo pulmonar.
La warfarina disminuye la protrombina y otros factores de procoagulación. Altera la vitamina K de un modo que reduce su capacidad para participar en la síntesis de los factores de coagulación dependientes de la vitamina K en el hígado. La warfarina se absorbe fácilmente después de la administración oral. Su efecto máximo toma 36 h a 72 h debido a las diferentes semividas de los factores de coagulación preformados que permanecen en la circulación.
La heparina se forma de modo natural y es liberada en cantidades pequeñas por los mastocitos en el tejido conectivo que rodea los capilares. Las preparaciones farmacológicas de heparina se extraen de tejidos animales. La heparina se une a antitrombina III, lo que causa un cambio de conformación que incrementa la capacidad de la anti-trombina III para inactivar trombina, factor Xa y otros factores de coagulación. Al promover la inactivación de los factores de coagulación, la heparina, por último, suprime la formación de fibrina.
La heparina es incapaz de cruzar las membranas del tubo digestivo y debe administrarse mediante inyección, casi siempre por infusión intravenosa. Las heparinas de bajo peso molecular se desarrollaron para inhibir la activación del factor X, pero tienen poco efecto en la trombina y otros factores de coagulación. Las heparinas de bajo peso molecular se administran por inyección subcutánea y requieren administración y vigilancia menos frecuente que la heparina estándar (no fraccionada).
Las complicaciones potenciales del consumo de warfarina son muchas. Además, la persona necesita someterse a pruebas de laboratorio frecuentes de su tiempo de anticoagulante con una prueba de la International Normalized Ratio (INR). Un nuevo anticoagulante oral que tiene menos complicaciones y requiere menos control es el dabigatrán, que se emplea de modo gradual en personas que tienen fibrilación auricular.
Los Centers for Disease Control (2010) estimaron que en 2050 habrá 12 millones de personas con fibrilación auricular. Por lo tanto, la aparición del dabigatrán es oportuna. Además, Freeman (2010) investigó el costo de utilizar dabigatrán en lugar de warfarina y halló que el dabigatrán es más efectivo en cuanto a costo cuando se emplea en personas con fibrilación auricular.
Retracción del coágulo
En condiciones normales, el coágulo se retrae dentro de los 20 min a 60 min que siguen a su formación, lo que contribuye a la hemostasia al exprimir el suero del coágulo y a unir los bordes del vaso roto. Las plaquetas, por efecto de la acción de sus filamentos de actina y miosina, contribuyen también a la retracción del coágulo y la hemostasia. La retracción del coágulo requiere grandes cantidades de plaquetas e indica un recuento bajo de plaquetas cuando no se efectúa.
Disolución del coágulo
La disolución de un coágulo de sangre comienza poco después de su formación. Esto permite que el flujo sanguíneo se restablezca y que la reparación tisular tenga lugar. El proceso por el cual se disuelve un coágulo de sangre se llama fibrinólisis. Así como la formación del coágulo, la disolución requiere una secuencia de pasos controlados por activadores e inhibidores. Elplasminógeno, la proenzima para el proceso fibrinolítico, normalmente se presenta en la sangre en su forma inactiva. Se convierte en su forma activa, la plasmina, mediante activadores de plasminógeno formados en el endotelio vascular, el hígado y los riñones. La plasmina formada de plasminógeno digiere las hebras de fibrina del coágulo y ciertos factores de coagulación, como fibrinógeno, factor V, factor VIII, protrombina y factor XII. La plasmina circulante es inactivada con rapidez por el inhibidor de α2-plasmina, que limita el proceso fibrinolítico al coágulo local y evita que se presente en la circulación completa.
Dos activadores de plasminógeno de origen natural son el activador de plasminógeno tipo tisular y el activador de plasminógeno tipo urocinasa. El hígado, el plasma y el endotelio vascular son las principales fuentes de activadores fisiológicos. Estos activadores se liberan en respuesta a diversos estímulos, incluidos fármacos vasoactivos, oclusión venosa, temperatura corporal alta y ejercicio. Los activadores son inestables e inactivados rápidamente por inhibidores sintetizados por el endotelio y el hígado. Por esta razón, la enfermedad hepática crónica puede alterar la actividad fibrinolítica.
Concentraciones altas de un inhibidor principal, el inhibidor activador de plasminógeno 1, se han relacionado con trombosis venosa profunda, enfermedad de la arteria coronaria e infarto de miocardio. Varios activadores de plasminógeno tisular (altepasa, reteplasa, tececteplasa), producidos mediante tecnología de ADN recombinante, están disponibles para el tratamiento de infarto de miocardio agudo, accidente cerebrovascular isquémico agudo y embolismo pulmonar.
Estados de hipercoagulabilidad
La hipercoagulabilidad constituye una forma exagerada de hemostasia que predispone a trombosis y oclusión de vasos sanguíneos. Hay 2 formas generales de estados de hipercoagulabilidad: afecciones que aumentan la función plaquetaria y afecciones que aceleran la actividad del sistema de coagulación. En el recuadro 26-1 se resumen los trastornos comúnmente relacionados con estados de hipercoagulabilidad. Los trombos arteriales suelen deberse a la turbulencia y se componen en gran parte de agregados plaquetarios. Por otro lado, los trombos venosos por lo general se deben a estasis del flujo y se componen en gran parte de agregados plaquetarios y complejos de fibrina resultantes de la activación de la cascada de coagulación.
Recuadro 26-1. Padecimientos relacionados con estados de hipercoagulabilidad:
- Incremento de la función plaquetaria
- Ateroesclerosis.
- Diabetes mellitus.Tabaquismo.
- Niveles altos de lípidos y colesterol en sangre.
- Aumento de las concentraciones de plaquetas.
- Actividad acelerada del sistema de coagulación
- Embarazo y puerperio.
- Consumo de anticonceptivos orales.
- Estado posquirúrgico.
- Inmovilidad.
- Insuficiencia cardíaca congestiva. Enfermedades malignas.
- El tabaquismo, las concentraciones sanguíneas altas de lípidos y colesterol, el estrés hemodinámico y la diabetes mellitus predisponen a lesión vascular, adherencia de plaquetas y, por último, trombosis.
Hipercoagulabilidad relacionada con función plaquetaria incrementada
La hipercoagulabilidad debida al aumento de la función plaquetaria produce adhesión de plaquetas, formación de coágulos de plaquetas e interrupción del flujo sanguíneo. Las causas de la función plaquetaria incrementada son perturbaciones del flujo, lesión endotelial y mayor sensibilidad de las plaquetas a factores que producen adhesividad y agregación. Las placas ateroescleróticas perturban el flujo de sangre al causar daño endotelial y promover la adherencia de plaquetas. Las plaquetas que se adhieren a la pared del vaso liberan factores de crecimiento, lo cual promueve la proliferación del músculo liso y, por lo tanto, contribuye al desarrollo de ateroesclerosis.
Trombocitosis
El término trombocitosis se utiliza para describir elevaciones en el recuento plaquetario por arriba de 1.000.000/μl. La trombocitosis puede presentarse como un proceso reactivo (trombocitosis secundaria) o como un proceso esencial (trombocitosis primaria).
Etiología y patogénesis
La trombopoyetina es la hormona clave en la regulación de la diferenciación de megacariocitos y la formación plaquetaria, aunque varias citocinas (ej. interleucina-6 e interleucina-11) también pueden desempeñar una función. Los megacariocitos y su progenie plaquetaria tienen receptores para trombopoyetina. La trombopoyetina se transporta en el plasma unida a receptores en la superficie de las plaquetas circulantes y en una forma no enlazada que está libre para promover la proliferación de megacariocitos. Cuando el recuento plaquetario baja, más trombopoyetina no unida está disponible para estimular la proliferación de megacariocitos y, si el recuento de plaquetas aumenta, menos trombopoyetina está disponible para estimular la proliferación. Así, en condiciones normales, la proliferación de megacariocitos y la producción de plaquetas están bajo el control de un mecanismo de retroalimentación negativa mediante el recuento de plaquetas.
La causa más común de trombocitosis secundaria es un estado de enfermedad que estimula la producción de trombopoyetina. El resultado es incremento de la proliferación de megacariocitos y la producción de plaquetas. Sin embargo, el recuento plaquetario pocas veces excede 1.000.000/μl. Las causas subyacentes más frecuentes de trombocitosis secundaria incluyen daño tisular secundario a intervención quirúrgica, infección, cáncer y padecimientos inflamatorios crónicos como artritis reumatoide y enfermedad de Crohn. Los únicos signos clínicamente evidentes suelen ser los de la enfermedad subyacente. La trombocitosis puede acompañar a otros trastornos mieloproliferativos, como policitemia vera y leucemia mixta.
La trombocitosis primaria o esencial es un trastorno mieloproliferativo (médula ósea) de las células madre hematopoyéticas. Aunque los niveles de trombopoyetina a menudo son normales en la trombocitosis esencial, las anomalías en el receptor de trombopoyetina y el enlace de plaquetas causan niveles más altos de lo esperado de trombopoyetina libre. Esto conduce a que la proliferación de megacariocitos y la producción de plaquetas aumenten. La disfunción de las plaquetas producidas contribuye a las principales características clínicas de hemorragia y trombosis.
Manifestaciones clínicas y tratamiento
Las manifestaciones clínicas habituales de la trombocitosis esencial son trombosis y hemorragia. Los fenómenos trombóticos incluyen trombosis venosaprofunda, embolismo pulmonar y trombosis de las venas porta y hepática. Algunas personas experimentan eritromelalgia, palpitación y ardor de los dedos causados por oclusión de las arteriolas por agregados plaquetarios. Por lo común, el trastorno se caracteriza por períodos asintomáticos largos interrumpidos por episodios trombóticos ocasionales y crisis hemorrágicas, que se presentan en personas con recuentos plaquetarios muy altos. El tratamiento comprende la administración de fármacos que disminuyen las plaquetas (ej. hidroxiurea) en casos de alto riesgo. El ácido acetilsalicílico puede ser un tratamiento complementario muy efectivo en individuos con complicaciones trombóticas recurrentes.
Hipercoagulabilidad relacionada con actividad de coagulación incrementada
La formación de trombos debida a la activación del sistema de coagulación puede ser resultado de trastornos primarios (genéticos) o secundarios (adquiridos) que afectan los componentes de coagulación del proceso de coagulación de la sangre (es decir, incremento de los factores procoagulantes o disminución de los factores anticoagulantes).
Trastornos heredados
De las causas heredadas de hipercoagulabilidad, las mutaciones en el gen del factor V y el gen de protrombina son las más frecuentes. Cerca del 2% al 15% de las personas caucásicas porta una mutación específica del factor V (denominada mutación de Leiden, en honor a la ciudad de Países Bajos donde se descubrió). En las personas con defectos heredados en el factor V, la proteína C no puede inactivar el factor mutante VA. En consecuencia, un importante mecanismo contrarregulador antitrombótico se pierde.
El defecto predispone a trombosis venosa. La frecuencia de la mutación puede ser de hasta el 60% entre las personas con trombosis venosa profunda recurrente.
Un simple cambio de nucleótidos en el gen de protrombina, que afecta del 1% al 2% de la población, se relaciona con altos niveles de protrombina y un incremento de casi el triple de las trombosis venosas. Los estados hipercoagulables primarios menos frecuentes incluyen las carencias heredadas de anticoagulantes como antitrombina III, proteína C y proteína S. Otro defecto hereditario produce concentraciones circulatorias altas de homocisteína, que predispone a trombosis venosa y arterial al activar las plaquetas y alterar los mecanismos antitrombóticos.
Trastornos adquiridos
Entre los factores adquiridos o secundarios que conducen a coagulación y trombosis incrementadasse encuentran estasis venosa por reposo prolongado en cama e inmovilidad, infarto al miocardio, cáncer, estados hiperestrogénicos y anticonceptivos orales. El tabaquismo y la obesidad promueven la hipercoagulabilidad por razones que se desconocen.
La estasis del flujo sanguíneo causa acumulación de factores de coagulación activados y plaquetas, y evita sus interacciones con inhibidores. El flujo lento y perturbado es una etiología común de trombosis venosa en personas inmovilizadas o postoperadas. Es importante asegurarse de que las personas inmovilizadas reciban tratamiento de heparina, si no está contraindicada, para prevenir la trombosis venosa profunda y el embolismo pulmonar. Además, la evidencia sugiere que los dispositivos de compresión neumática intermitente son muy útiles para evitar complicaciones tromboembólicas con la inmovilidad.
Las personas con enfermedad inflamatoria intestinal están en alto riesgo de tromboembolismo venoso y arterial, pero también pueden sufrir efectos adversos, como hemorragia gastrointestinal, si se les administran anticoagulantes. Por lo tanto, es importante minimizar los factores que promueven efectos trombóticos, como inflamación, tratamiento con esteroides, hospitalización, anticoncepción oral, insuficiencia de vitaminas B y folato, tabaquismo y empleo de catéteres intravenosos centrales con el fin de que se requiera poca o ninguna anticoagulación. La insuficiencia cardíaca también contribuye a congestión venosa y trombosis.
Los síndromes de hiperviscosidad (policitemia) y los eritrocitos deformados en la enfermedad de células falciformes incrementan la resistencia al flujo y causan estasis de vasos pequeños.
La incidencia de accidente cerebrovascular, tromboémbolos e infarto al miocardio es mayor en mujeres que utilizan anticonceptivos orales, en particular en las mayores de 35 años de edad y las que fuman mucho. Los factores de coagulación se incrementan también durante el embarazo normal.
Estos cambios, junto con la actividad limitada durante el puerperio (período posparto inmediato), predisponen a trombosis venosa.
Asimismo, la hipercoagulabilidad es común en el cáncer y la septicemia. Se cree que muchas células tumorales liberan moléculas de factor tisular, que, junto con el incremento de la inmovilidad y la septicemia que se observan en personas con enfermedad maligna, contribuyen a la trombosis en estas personas.
Síndrome antifosfolípido
Otra causa de trombosis venosa y arterial incrementada es el síndrome antifosfolípido. Esta afección se relaciona con autoanticuerpos (sobre todo inmunoglobulina G [IgG]) dirigidos contra fosfolípidos de enlace a proteínas, cuyo resultado es aumento de la actividad de coagulación. Las características comunes del síndrome antifosfolípido son trombos venosos y arteriales, pérdida fetal recurrente y trombocitopenia. El trastorno puede ser un padecimiento primario que se presenta aislado con signos de hipercoagulabilidad o una afección secundaria algunas veces relacionada son lupus eritematoso sistémico.
Etiología y patogénesis
Aunque los mecanismos para este síndrome se desconocen, se identifican varias vías posibles:
- Es posible que los anticuerpos interfieran en la cascada de coagulación y conduzcan a un estado de hipercoagulabilidad.
- Los anticuerpos podrían unirse directamente a la superficie celular endotelial y causar secreción de citocinas que producen activación y agregación plaquetarias.
- Los anticuerpos pueden dirigirse a una proteína sérica de enlace a fosfolípidos que funciona como un anticoagulante.
Además de la acción de los anticuerpos, parece probable que otros factores desempeñen la función de determinar si una persona presenta manifestaciones clínicas del trastorno. Aunque sólo se especula, estos factores podrían incluir traumatismo vascular o la presencia de infección que da lugar a la producción de citocina y la activación de células endoteliales.
Manifestaciones clínicas
Las personas con la enfermedad presentan diversas manifestaciones clínicas, por lo general, las que se caracterizan por trombos venosos y arteriales recurrentes.
También podrían ocurrir vegetaciones valvulares cardíacas relacionadas con la adhesión de trombos y trombocitopenia debida al consumo excesivo de plaquetas. La trombosis venosa, sobre todo en las venas profundas de las piernas, se observa hasta en el 50% de las personas con el síndrome y la mitad de ellas desarrolla embolismo pulmonar. La trombosis arterial afecta el cerebro hasta en el 50% de los casos y causa ataques isquémicos transitorios o accidentes cerebrovasculares. Otros sitios de trombosis arterial son las arterias coronarias del corazón y las arterias retinanina, renal y periférica.
Las mujeres con el trastorno por lo común tienen antecedentes de pérdidas de embarazo recurrentes por isquemia y trombosis de los vasos placentarios. Estas mujeres tienen también mayor riesgo de dar a luz a un lactante prematuro debido a hipertensión relacionada con el embarazo e insuficiencia uteroplacentaria.
En la mayoría de las personas con síndrome antifosfolípido, los accidentes trombóticos se presentan con un solo episodio en un sitio anatómico. Meses o años más tarde, algunas personas pueden tener recurrencias que imitan al accidente inicial. Ocasionalmente, alguien tiene múltiples oclusiones vasculares relacionadas con muchos sistemas orgánicos. Esta afección de inicio rápido se denomina síndrome antifosfolípido catastrófico y su tasa de mortalidad es alta.
Tratamiento
El tratamiento del síndrome se enfoca en suprimir o reducir los factores que predisponen a trombosis, incluidas la recomendación de abandonar el tabaquismo y asesoría contra el consumo de anticonceptivos orales que contienen estrógeno. El accidente trombótico agudo se trata con anticoagulantes (heparina y warfarina) y supresión inmunitaria en casos refractarios. Puede consumirse ácido acetilsalicílico y fármacos anticoagulantes para prevenir trombosis futuras.
Trastornos hemorrágicos
Los trastornos hemorrágicos o el deterioro de la coagulación sanguínea pueden deberse a defectos en alguno de los factores que contribuyen a la hemostasia. Es posible que la hemorragia sea resultado de trastornos relacionados con el número o la función de las plaquetas, factores de coagulación e integridad de los vasos sanguíneos.
Hemorragia relacionada con trastornos plaquetarios
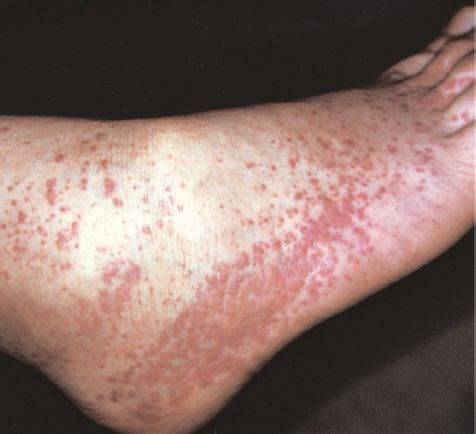
La hemorragia debida a trastornos plaquetarios refleja un disminución de la cantidad de plaquetas secundaria a la producción reducida, destrucción incrementada o función deteriorada. La hemorragia espontánea de trastornos plaquetarios con mucha frecuencia se relaciona con vasos pequeños de las membranas mucosas y la piel. Los sitios frecuentes de hemorragia son las membranas mucosas de nariz, boca, tubo digestivo y cavidad uterina. La hemorragia cutánea se ve como pequeñas hemorragias (petequias) y zonas púrpuras de magulladuras (púrpura) en áreas dependientes en las que la presión capilar es mayor (figura 26-4). Las petequias se ven casi exclusivamente en el padecimiento de insuficiencia plaquetaria y no de disfunción. La hemorragia de vasos intracraneales es un peligro raro con agotamiento plaquetario grave.
Trombocitopenia
Una reducción del número de plaquetas, también conocida como trombocitopenia, es una causa importante de hemorragia generalizada. Trombocitopenia suele referirse a disminución del número de plaquetas circulantes a un nivel menor de 150.000/μl. Entre mayor sea la disminución del recuento de plaquetas, mayor será el riesgo de hemorragia. La trombocitopenia puede ser consecuencia de descenso de la producción de plaquetas, aumento del secuestro de plaquetas en el bazo o reducción de la supervivencia de plaquetas.
La disminución de la producción de plaquetas por pérdida de la función de la médula ósea tiene lugar en la anemia aplásica. El reemplazo de médula ósea por células malignas, como se observa en la leucemia, también ocasiona una menor producción de plaquetas. La radioterapia y fármacos como los que se emplean en el tratamiento del cáncer pueden deprimir la función de la médula ósea y disminuir la producción plaquetaria. Es posible que la infección con virus de inmunodeficiencia humana (VIH) o citomegalovirus suprima la producción de megacariocitos, los precursores de plaquetas.
La producción de plaquetas puede ser normal, pero es factible que se acumulen de modo excesivo en el bazo. Aunque el bazo normalmente secuestra del 30% al 40% de las plaquetas antes de liberarlas a la circulación, la proporción puede ser de hasta el 90% cuando se agranda en la esplenomegalia. Si es necesario, la trombocitopenia hiperesplénica puede tratarse con esplenectomía.
La reducción de la supervivencia plaquetaria se debe a diversos mecanismos inmunitarios y autoinmunitarios. Anticuerpos antiplaquetarios pueden destruir plaquetas. Los anticuerpos pueden dirigirse contra los autoantígenos plaquetarios o contra antígenos en las plaquetas de transfusiones sanguíneas o embarazo. Los anticuerpos se dirigen a las glucoproteínas de la membrana plaquetaria GPIIb/IIIa y GPIb/IX. La destrucción no inmunitaria de plaquetas es consecuencia de lesión mecánica por válvulas cardíacas prostéticas o hipertensión maligna, que estrechan vasos pequeños. En CID aguda o púrpura trombocitopénica trombótica, el consumo excesivo de plaquetas da lugar a insuficiencia.
Trombocitopenia inducida por fármacos
Algunos fármacos, como quinina, quinidina y ciertos antibióticos que contienen sulfas, pueden inducir la trombocitopenia. Estos medicamentos inducen una respuesta antígeno-anticuerpo y la formación de complejos inmunes que ocasionan destrucción de plaquetas por lisis mediada por complemento. Las personas con trombocitopenia relacionada con fármacos experimentan una caída rápida del recuento plaquetario de 2 a 3 días de volver a tomar el medicamento o 7 o más días (es decir, el tiempo necesario para preparar una respuesta inmunitaria) después de empezar uno por primera vez. El recuento plaquetario aumenta con rapidez tras descontinuar el fármaco.
Trombocitopenia inducida por heparina
La trombocitopenia inducida por heparina (TIH) se relaciona con la heparina farmacológica anticoagulante. El 10% de las personas tratadas con heparinamanifiesta trombocitopenia transitoria leve en 2 a 5 días de empezar el fármaco. Sin embargo, alrededor del 1% al 5% de quienes reciben heparina sufre accidentes trombóticos que ponen en riesgo la vida 1 a 2 semanas después de iniciar el tratamiento. La TIH se debe a una reacción inmunitaria dirigida contra un complejo de heparina y factor plaquetario 4, un componente normal de los gránulos plaquetarios que se une con firmeza a la heparina. El enlace de un anticuerpo con el factor plaquetario produce complejos inmunitarios que activan las plaquetas restantes, lo que da lugar a trombosis. Además, las partículas de plaquetas protrombóticas y la inducción del factor tisular continúan promoviendo la coagulación.
El tratamiento de la TIH requiere descontinuar de inmediato el tratamiento con heparina y el consumo alternativo de anticoagulantes para evitar que la trombosis recurra. Se ha demostrado que la heparina más reciente de peso molecular bajo es efectiva para reducir la incidencia de complicaciones inducidas por heparina en comparación con la forma anterior del fármaco de alto peso molecular.
Púrpura trombocitopénica inmunitaria
La púrpura trombocitopénica inmunitaria (PTI) es una enfermedad autoinmunitaria que causa formación de anticuerpos plaquetarios y destrucción excesiva de plaquetas. La enfermedad puede tener lugar en ausencia de cualquier factor de riesgo conocido (PTI primaria o idiopática) o como un trastorno secundario debido a una alteración subyacente y como una enfermedad aguda (duración de 6 meses o menos) o crónica. Algunas formas secundarias de PTI se relacionan con el síndrome de inmunodeficiencia adquirida (sida), lupus eritematoso sistémico, síndrome antifosfolípido, leucemia linfocítica crónica, linfoma, hepatitis C y medicamentos como heparina y quinidina.
Un trastorno de PTI aguda ocurre en niños pequeños (5 años de edad) y, por lo general, sigue a una infección viral. Se caracteriza por inicio repentino de petequias y púrpura, y suele ser un trastorno autolimitado que no requiere tratamiento. La mayoría de niños se recupera en pocas semanas. En contraste, la PTI primaria a menudo es un trastorno crónico en adultos con un inicio insidioso que pocas veces sigue a una infección.
Etiología y patogénesis
Se cree que la trombocitopenia que acompaña al PTI es resultado de múltiples mecanismos que incluyen anticuerpos antiplaquetarios contra glucoproteínas (IIb/IIIa y Ib/IX) en la membrana plaquetaria. Las plaquetas, que se hacen más susceptibles a la fagocitosis por el anticuerpo, se destruyen en el bazo. Los niveles plasmáticos de trombopoyetina, el factor principal que estimula el crecimiento y desarrollo de megacariocitos, no son altos en personas con PTI. Las pruebas indican que el PTI es causado por disfunción de células T, en específico células reguladoras CD4 y T, que activan la respuesta inmunitaria y pasan a trombocitopenia.
Manifestaciones clínicas
Las manifestaciones de PTI incluyen antecedentes de equimosis, sangrado de encías, epistaxis (es decir, hemorragia nasal), melena y hemorragia menstrual anómala en quienes tienen recuento plaquetario moderadamente reducido. Ya que el bazo es el sitio donde las plaquetas se destruyen, puede observarse agrandamiento esplénico. La afección se descubre de modo incidental o como resultado de signos de hemorragia, a menudo hacia la piel (es decir, púrpura y petequias) o la mucosa bucal.
Diagnóstico y tratamiento
El diagnóstico de la PTI suele basarse en trombocitopenia grave (recuento plaquetario <20.000/μl a 30.000/μl) y exclusión de otras causas. Existen pruebas para los anticuerpos unidos a plaquetas, pero carecen de especificidad (ej. reaccionan con anticuerpos plaquetarios de otras fuentes). La forma secundaria de PTI a veces imita la forma idiopática del trastorno, por lo tanto, el diagnóstico se establece sólo después de excluir otras causas conocidas de trombocitopenia.
La decisión de tratar la PTI se basa en el recuento de plaquetas y el grado de hemorragia. Muchas personas con PTI están bien sin tratamiento. Los corticoesteroides casi siempre se utilizan como tratamiento inicial. Otros tratamientos iniciales efectivos incluyen globulina inmune intravenosa. Sin embargo, este tratamiento es costoso y el efecto beneficioso dura sólo 1 a 2 semanas.
Púrpura trombocitopénica trombótica
La púrpura trombocitopénica trombótica (PTT) es una combinación de trombocitopenia, anemia hemolítica, insuficiencia renal, fiebre y anomalías neurológicas. Es un trastorno raro que probablemente resulta de la introducción de sustancias de agregación plaquetaria a la circulación. La etiología subyacente de muchos casos es la insuficiencia de una enzima (designada ADAMTS 13, por sus siglas en inglés) que degrada multímeros de peso molecular grande de FvW, lo que permite que se acumulen y causen agregación plaquetaria y adhesión al endotelio.
Etiología y patogénesis
Es posible que la insuficiencia enzimática sea heredada o adquirida como una consecuencia de anticuerpos dirigidos contra la enzima. La PTT suele afectar a personas previamente sanas, pero también se relaciona con enfermedades del colágeno autoinmunitarias, fármacos, infecciones como VIH y embarazo. El trastorno es similar a la CID, pero no se vincula con el sistema de coagulación. Las toxinas producidas por algunas cepas de Escherichia coli (ej. E. coli O157: H7) causan lesión endotelial y producen una afección similar, el síndrome urémico hemolítico (SUH).
El inicio de la PTT es abrupto y el resultado final puede ser fatal. Las oclusiones vasculares diseminadas se deben a trombos en arteriolas y capilares de muchos órganos, incluido corazón, cerebro y riñones. Los eritrocitos se fragmentan conforme circulan por los vasos parcialmente ocluidos, lo que ocasiona anemia e ictericia.
Manifestaciones clínicas y tratamiento
Las manifestaciones clínicas comprenden púrpura, petequias, hemorragia vaginal y síntomas neurológicos que van desde cefalea hasta convulsiones y conciencia alterada. El tratamiento de urgencia para la PTT implica plasmaféresis, un procedimiento que conlleva la remoción de plasma de la sangre extraída y reemplazo con plasma recién congelado.
La infusión de plasma proporciona la enzima insuficiente. Con la plasmaféresis y el tratamiento de infusión de plasma, la recuperación es completa en 80% de los casos.
Función plaquetaria deteriorada
El trastorno de la función plaquetaria (denominado también trombocitopatía) puede deberse a enfermedades de adhesión heredadas (ej. enfermedad de von Willebrand) o defectos adquiridos ocasionados por fármacos, enfermedad o intervención quirúrgica que requiere circulación extracorpórea (es decir, derivación cardiopulmonar). La función plaquetaria insuficiente también es común en la uremia, tal vez como resultado de productos de desecho no excretados.
La administración de ácido acetilsalicílico y otros medicamentos antiinflamatorios no esteroideos (AINE) es la causa más frecuente de disfunción plaquetaria. El ácido acetilsalicílico produce acetilación irreversible de la actividad de la ciclooxigenasa plaquetaria y, en consecuencia, la síntesis de TXA2 necesaria para la agregación plaquetaria. En contraste con los efectos del ácido acetilsalicílico, la inhibición de ciclooxigenasa por otros AINE es reversible y dura sólo el tiempo que la acción del fármaco persiste. Se recurre al ácido acetilsalicílico (81 mg al día) para evitar la formación de trombos arteriales y reducir el riesgo de ataque cardíaco y accidente cerebrovascular. En el recuadro 26-2 se enumeran otros medicamentos que afectan la función plaquetaria.
Recuadro 26-2. Fármacos que pueden predisponer a hemorragia (la lista no es exhaustiva):
- Interferencia con la producción o función plaquetarias
- Acetazolamida.
- Antimetabolitos y fármacos anticáncer.
- Antibióticos, como penicilina y cefalosporinas.
- Ácido acetilsalicílico y salicilatos.
- Carbamacepina.
- Clofibrato.
- Colchicina.
- Dipiradamol.
- Diuréticos tiacídicos.
- Sales de oro.
- Heparina.
- AINE.
- Derivados de quinina (quinidina e hidroxicloroquina).
- Sulfonamidas.
- Interferencia con factores de coagulación
- Amiodarona.
- Esteroides anabólicos.
- Warfarina.
- Heparina.
- Disminución de las concentraciones de vitamina K
- Antibióticos.
- Clofibrato.
Hemorragia relacionada con insuficiencias del factor de coagulación
Los defectos de coagulación sanguínea pueden ser consecuencia de insuficiencia o función alterada de uno o más de los factores de coagulación, incluido el FvW. Las insuficiencias surgen como resultado de enfermedad heredada o síntesis defectuosa o incremento del consumo de factores de coagulación. La hemorragia secundaria a las insuficiencias del factor de coagulación suelen tener lugar después de lesión o traumatismo. Son comunes las magulladuras grandes, hematomas y hemorragia prolongada hacia el tracto gastrointestinal, el urinario o las articulaciones.
Trastornos heredados
La enfermedad de von Willebrand y la hemofilia (A y B) son 2 de los trastornos hemorrágicos heredados más frecuentes. La enfermedad de von Willebrand se considera la coagulopatía here-dada más común y afecta a cerca del 1% al 2% de la población. La hemofilia A (insuficiencia del factor VIII ) afecta a 1 de 5.000 nacimientos vivos de varones. La hemofilia B (insuficiencia del factor ix) ocurre en alrededor de 1 de 20.000 personas, lo que explica 15% de personas con hemofilia. Es genética y clínicamente similar a la hemofilia A. La enfermedad de von Willebrand y la hemofilia A se deben a defectos relacionados con el complejo del factor VIII-FvW. El FvW, que es sintetizado por el endotelio y los megacariociotos, se requiere para la adhesión de plaquetas a la matriz subendotelial del vaso sanguíneo. Sirve también como portador del factor VIII y es importante para la estabilidad del factor VIII en la circulación al prevenir su proteólisis. El hígado y las células endoteliales producen la proteína coagulante de la porción funcional del factor VIII. Así, los factores VIII y FvW, que se sintetizan por separado, entran juntos al plasma y circulan en él como una unidad que promueve la coagulación y la adhesión plaquetaria a la pared del vaso.
Enfermedad de von Willebrand
La enfermedad de von Willebrand es un trastorno hemorrágico hereditario frecuente que se caracteriza por una insuficiencia o un defecto en FvW. Se han descrito hasta 20 variantes de la enfermedad de von Willebrand. Estas variantes pueden agruparse en 2 categorías: tipos 1 y 3, que se relacionan con bajos niveles de FvW y tipo 2, que se distingue por defectos en el FvW.
Clasificación
El tipo 1, un trastorno autosómico dominante, constituye cerca de 70% de los casos y es relativamente leve. El tipo 2, también un trastorno autosómico dominante, explica cerca del 25% de los casos y se relaciona con hemorragia leve a moderada. El tipo 3, que es un trastorno autosómico recesivo más o menos raro, se relaciona con niveles en extremo bajos de FvW funcional y, por consiguiente, manifestaciones clínicas graves. Las personas con enfermedad de von Willebrand tienen un defecto compuesto que compromete la función plaquetaria y la vía de la coagulación.
Manifestaciones clínicas
Incluyen hemorragia espontánea nasal, bucal y del tubo digestivo, flujo menstrual excesivo y tiempo de hemorragia prolongado en presencia de un recuento plaquetario normal. Casi todos los casos (es decir, los tipos 1 y 2) son leves y no requieren tratamiento, y muchas personas con el trastorno se diagnostican cuando una intervención quirúrgica o extracción dental produce hemorragia prolongada. En casos graves (es decir, tipo 3), la hemorragia gastrointestinal que pone en riesgo la vida y la hemorragia de las articulaciones pueden ser similares a la que se observa en la hemofilia. La hemorragia relacionada con la enfermedad de von Willebrand suele ser leve y no se administra ningún tratamiento sistemático distinto a la evasión del ácido acetilsalicílico.
Hemofilia A
La hemofilia A es un trastorno recesivo ligado a X que afecta sobre todo a los varones.Aunque es hereditario, no hay antecedentes familiares del trastorno en casi el 30% de los casos recién diagnosticados, lo que sugiere que surgió como una nueva mutación en el gen del factor VIII4.
Alrededor del 90% de las personas con hemofilia produce cantidades insuficientes del factor y 10% produce una forma defectuosa. El porcentaje de actividad normal del factor VIII en la circulación depende del defecto genético y determina la gravedad de la hemofilia (es decir, del 6% al 30% en la hemofilia leve, del 2% al 5% en la hemofilia moderada y el 1% o menos en formas graves de hemofilia). En las formas leve o moderada de la enfermedad, la hemorragia no suele presentarse a menos que haya una lesión local o traumatismo como intervención quirúrgica o procedimiento dental. Es posible que el trastorno leve no se detecte en la infancia. En la hemofilia grave, la hemorragia casi siempre ocurre en la infancia (ej. puede advertirse en el momento de la circuncisión), es espontánea, grave y a menudo se presenta varias veces al mes.
Manifestaciones clínicas
De manera característica, la hemorragia tiene lugar en tejidos blandos, tubo digestivo y cadera, rodilla, codo y articulaciones del tobillo. La hemorragia espontánea de las articulaciones suele iniciar cuando un niño comienza a caminar. Con frecuencia, la articulación objetivo es propensa a hemorragia repetida. La hemorragia causa inflamación de la membrana sinovial, con dolor agudo e hinchazón. Sin el tratamiento adecuado, la hemorragia crónica y la inflamación producen fibrosis articular y contracturas, cuyo resultado es incapacidad mayor.
Tratamiento
La prevención del traumatismo es importante en personas con hemofilia. Debe evitarse el ácido acetilsalicílico y otros AINE que afectan la función plaquetaria. El tratamiento de restitución del factor VIII que se administra en casa reduce el daño musculoesquelético característico. Se inicia cuando la hemorragia se presenta o como profilaxis con episodios hemorrágicos repetidos. Es posible que los productos recombinantes y las bombas de infusión continua sean preventivos más que terapéuticos. El desarrollo de anticuerpos inhibidores para el factor recombinante VIII aún es una complicación importante del tratamiento.
La clonación del gen del factor VIII y el avance en los sistemas de creación de genes son esperanzadores con respecto a que la hemofilia A pueda curarse con el tratamiento de restitución génica. La detección del portador y el diagnóstico parental pueden realizarse ahora mediante análisis de mutación directa de genes o estudios de enlace de ADN. La amniocentesis prenatal o muestreo de vellosidades coriónicas se utiliza para predecir complicaciones y determinar el tratamiento. Por último, puede emplearse para seleccionar a pacientes para la adición de genes.
Trastornos adquiridos
Los factores de coagulación V, VII, IX, X, XI y XII, la protrombina y el fibrinógeno se sintetizan en el hígado. En la enfermedad hepática, la síntesis de factores de coagulación se reduce, lo que podría producir hemorragia. De los factores de coagulación que se sintetizan en el hígado, los factores II, VII, IX y, X, y la protrombina requieren la presencia de vitamina K para su actividad normal. En la insuficiencia de vitamina K, el hígado produce el factor de coagulación, pero en una forma inactiva.
La vitamina K es una vitamina liposoluble que bacterias intestinales sintetizan de manera continua. Esto significa que la insuficiencia de vitamina K es improbable a menos que la síntesis intestinal se interrumpa o la absorción de la vitamina se afecte. La insuficiencia de vitamina K puede presentarse en el neonato antes que la flora intestinal se establezca. También puede ocurrir como resultado del tratamiento con antibióticos de amplio espectro que destruyen la flora intestinal. Como la vitamina K es liposoluble, su absorción requiere sales biliares. Es posible que la insuficiencia de vitamina K sea consecuencia de absorción alterada de grasa secundaria a la enfermedad hepática o de la vesícula biliar.
Hemorragia relacionada con trastornos vasculares
La hemorragia por trastornos vasculares a veces se denomina púrpura no trombocitopénica. Estos trastornos pueden ser el resultado de paredes vasculares estructuralmente débiles o de daño de los vasos por inflamación o respuestas inmunitarias. Muy a menudo se caracterizan por equimosis fácil, aparición espontánea de petequias y púrpura en piel, y membranas mucosas. El recuento plaquetario y los resultados de otras pruebas de factores de coagulación son normales en personas con trastornos hemorrágicos causados por defectos vasculares.
Entre los trastornos vasculares que causan hemorragia están la telangiectasia hemorrágica, un trastorno autosómico frecuente caracterizado por capilares dilatados de pared delgada y arteriolas; insuficiencia de vitamina C (es decir, escorbuto), que conduce a síntesis insuficiente de colágeno y fallo de las células endoteliales para cementarse juntas de forma adecuada, lo que hace que la pared vascular sea frágil; enfermedad de Cushing, que causa atrofia proteínica y pérdida de soporte del tejido vascular por exceso de cortisol; y púrpura senil (es decir, equimosis en adultos mayores), ocasionada por deterioro de la síntesis de colágeno en el proceso de envejecimiento. Los defectos vasculares también tienen lugar en el curso de la CID o como consecuencia de microtrombos y tratamiento con costicoesteroides.
Coagulación intravascular diseminada
La CID es una paradoja en la secuencia hemostática y se caracteriza por coagulación generalizada y hemorragia en el compartimiento vascular. No es una enfermedad primaria pero se observa como una complicación de una amplia variedad de afecciones. La CID comienza con activación masiva de la secuencia de coagulación por la generación no regulada de trombina, que conduce a la formación sistémica de fibrina. Además, los niveles de todos los anticoagulantes principales se reducen (figura 26-5).
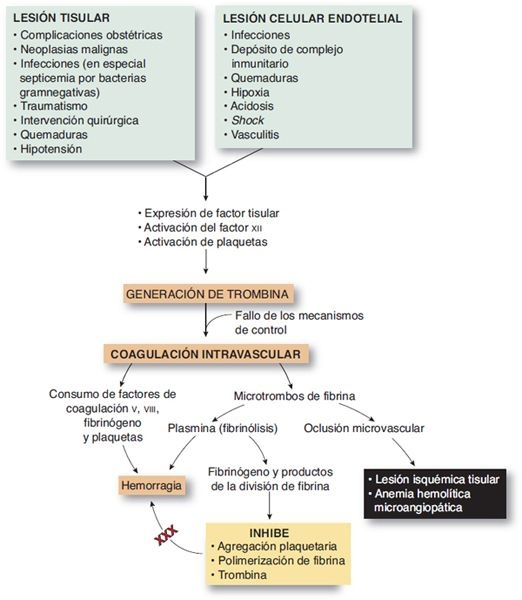
Los microtrombos resultantes causan oclusión vascular e isquemia tisular. La insuficiencia orgánica múltiple es posible. La formación del coágulo consume todas las proteínas de coagulación y plaquetas, lo que produce hemorragia grave.
Etiología y patogénesis
El trastorno puede iniciarse por la activación de la vía intrínseca, de la extrínseca o de ambas. La activación por la vía extrínseca tiene lugar con liberación de factores tisulares y se relaciona con complicaciones obstétricas, traumatismo, septicemia bacteriana y cánceres.
La vía intrínseca se puede activar mediante daño endotelial extenso, con activación del factor XII. La lesión endotelial puede ser causada por virus, infecciones, mecanismos inmunitarios, estasis de la sangre o extremos de temperatura. Las vías de anti-coagulación afectadas se vinculan también con niveles reducidos de antitrombina y el sistema anticoagulante de proteína C en la CID.
Hay evidencias de que la causa subyacente de la CID es la infección o inflamación y las citocinas (factor de necrosis tumoral, interleucina-1 y otros) que se libera en el proceso son los mediadores desencadenantes. Estas citocinas no sólo median la inflamación, sino también pueden incrementar la expresión del factor tisular en las células endoteliales y de modo simultáneo disminuir la expresión de trombomodulina. La trombomodulina, una glucoproteína presente en la membrana celular de las células endoteliales, se une a trombina y actúa como un mecanismo regulador adicional en la coagulación. Los padecimientos clínicos comunes que pueden causar CID incluyen trastornosobstétricos, que explican el 50% de los casos, traumatismo masivo, shock, septicemia y enfermedad maligna. En el recuadro 26-3 se resumen las afecciones relacionadas con CID.
Los factores que participan en los padecimientos que causan CID a menudo están interrelacionados. En complicaciones obstétricas, los factores tisulares liberados de líquido placentario necrótico o líquido tisular fetal o amniótico podrían desencadenar la CID. La hipoxia, el estado de shock y la acidosis que pueden coexistir contribuyen también al causar lesión endotelial.
Las infecciones por bacterias gramnegativas dan como resultado liberación de endotoxinas, que activan la vía extrínseca por liberación del factor tisular y la vía intrínseca por daño endotelial. Las endotoxinas inhiben también la actividad de la proteína C.
Recuadro 26-3. Padecimientos que se han relacionado con CID:
- Padecimientos obstétricos
- Desprendimiento de placenta.
- Síndrome de feto muerto.
- Preeclampsia y eclampsia.
- Embolia del líquido amniótico.
- Cánceres
- Cáncer metastático.
- Leucemia.
- Infecciones
- Infecciones bacterianas agudas (ej. meningitis meningocócica).
- Infecciones virales agudas.
- Infecciones por rickettsias (ej. fiebre manchada de las Montañas Rocosas).
- Infecciones parasitarias (ej. paludismo).
- Shock
- Shock séptico.
- Shock hipovolémico grave.
- Traumatismo e intervención quirúrgica
- Quemaduras.
- Traumatismo masivo.
- Circulación extracorpórea relacionada con intervención quirúrgica.
- Mordedura de serpiente.
- Golpe de calor.
- Padecimientos hemorrágicos
- Reacciones a transfusión sanguínea.
Manifestaciones clínicas
Aunque la coagulación y la formación de microémbolos caracterizan la CID, por lo general, susmanifestaciones agudas se relacionan de manera más directa con los problemas hemorrágicos que la acompañan. La hemorragia puede evidenciarse como petequias, púrpura, rezuma de sitios de punción o hemorragia grave.
La hemorragia posparto no controlada puede indicar CID. Es posible que los microémbolos obstruyan vasos sanguíneos y causen hipoxia tisular y daño necrótico a estructuras orgánicas, como riñones, corazón, pulmones y cerebro. En consecuencia, los signos clínicos comunes pueden deberse a insuficiencia renal, circulatoria o respiratoria, úlceras hemorrágicas agudas o convulsiones y coma. Una forma de anemia hemolítica puede presentarse cuando los eritrocitos se lesionan al pasar por vasos parcialmente bloqueados por trombos.
Tratamiento
El tratamiento de la CID se dirige a controlar la enfermedad primaria, restituir componentes de la coagulación y evitar la activación posterior de mecanismos de coagulación. Las transfusiones de plasma congelado fresco, plaquetas o crioprecipitado que contiene fibrinógeno podrían corregir la insuficiencia del factor de coagulación.