01. Mecanismos de la Hemostasia
Mecanismos de la hemostasia
La hemostasia se divide en 3 etapas:
- Constricción vascular.
- Formación del tapón plaquetario.
- Coagulación sanguínea (figura 26-1).
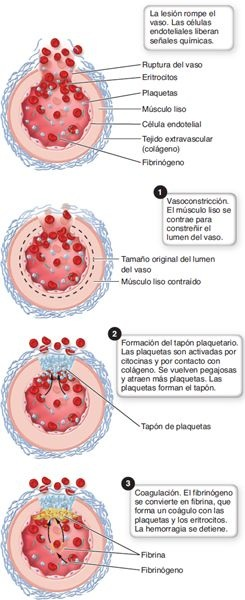
Durante el proceso de hemostasia, las hebras de fibrina similares al pelo pegan las plaquetas agregadas para formar la base estructural del coágulo sanguíneo. En presencia de fibrina, el plasma se vuelve gelatinoso y atrapa eritrocitos y otros elementos formados en la sangre. La hemostasia se completa cuando el tejido fibroso crece hacia el coágulo y sella el agujero en el vaso.
Constricción vascular
El espasmo de los vasos constriñe el vaso y reduce el flujo sanguíneo. Es un fenómeno transitorio que suele durar minutos u horas. El espasmo de los vasos se inicia por lesión endotelial y es causado por mecanismos locales y humorales. Los reflejos neurales y el tromboxano A (TXA de las plaquetas y otros mediadores, como la serotonina, contri-2), una prostaglandina liberada de las plaquetas y otros mediadores, como la serotonina, contribuyen a la vasoconstricción. El vasoconstrictor más poderoso es la endotelina. La prostaciclina, otra prostaglandina liberada del endotelio del vaso, produce vasodilatación e inhibe la agregación plaquetaria en el endotelio no lesionado circundante.
Formación del tapón plaquetario
El tapón plaquetario, la segunda línea de defensa, se inicia cuando las plaquetas entran en contacto con la pared vascular. Pequeñas rupturas en la pared del vaso a menudo se sellan con el tapón plaquetario y no con un coágulo de sangre.
Las plaquetas, o trombocitos, son grandes fragmentos del citoplasma de células de la médulaósea llamados megacariocitos.
La plaqueta tiene una vida media cercana a 8 a 12 días y luego los macrófagos se encargan de descomponerla y eliminarla. La concentración sérica normal se aproxima a 150.000 a 400.000 plaquetas por microlitro (μl) de sangre. La producción de plaquetas está controlada por una proteína, llamada trombopoyetina, que causa proliferación y maduración de megacariocitos. Las fuentes de trombopoyetina incluyen hígado, riñones, músculo liso y médula ósea.
Las plaquetas, poseen una membrana celular pero no núcleo y no pueden reproducirse. La membrana celular tiene fosfolípidos que ayudan en el proceso de coagulación. Aunque carecen de un núcleo, tienen muchas de las características de una célula completa. La membrana celular externa está cubierta por una capa de glucoproteínas, glucosaminoglucanos y proteínas de coagulación (figura 26-2).
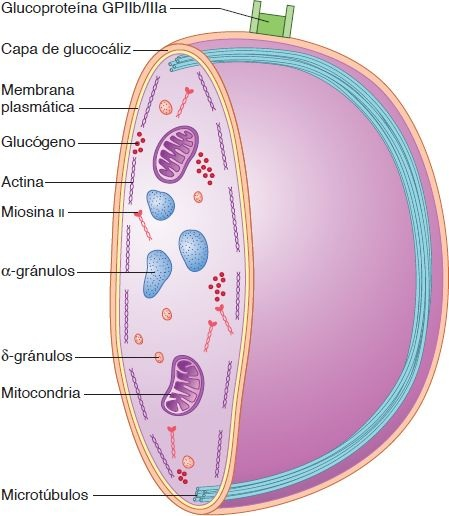
Una de las glucoproteínas importantes es GPIIb/IIIa, que se une al fibrinógeno y une las plaquetas entre sí. Microtúbulos y filamentos de actina y miosina que soportan la membrana celular mantienen la forma de la plaqueta. Las plaquetas tienen mitocondrias y sistemas enzimáticos capaces de producir trifosfato de adenosina (ATP) y difosfato de adenosina (ADP). Poseen también las enzimas necesarias para la síntesis de la prostaglandina, TXA2, que se requiere para su función en la hemostasia.
Las plaquetas tienen 2 tipos específicos de gránulos (α y θ) que liberan mediadores para la hemostasia. Los α-gránulos expresan selectina P, una proteína adhesiva en su superficie y contienen fibrinógeno, factor de von Willebrand (FvW), fibronectina, factores V y VIII, factor plaquetario (una quimiocina que se une a heparina), factor de crecimiento derivado de plaquetas (FCDP), factor de crecimiento transformador (FCT-α) y trombospondina. La liberación de factores de crecimiento da como resultado la proliferación y crecimiento de células endoteliales vasculares, células de músculo liso y fibroblastos, y es importante en la reparación vascular. Los θ-gránulos, o gránulos densos, contienen ADP y ATP, calcio ionizado, histamina, serotonina y adrenalina, los cuales contribuyen a la vasoconstricción.
La formación del tapón plaquetario implica adhesión y agregación de plaquetas. Las plaquetas son atraídas a la pared del vaso dañado, se activan y cambian de discos lisos a esferas espinosas, con lo que exponen los receptores de glucoproteína en sus superficies. La adhesión de plaquetas requiere una molécula de proteína llamada factor de von Willebrand, que se fuga hacia el tejido lesionado desde el plasma. Este factor es producido por las células endoteliales de los vasos sanguíneos y circula en la sangre como una proteína portadora para el factor de coagulación VIII. La adhesión a la capa subendotelial del vaso tiene lugar cuando el receptor de plaquetas se une al FvW en el sitio de la lesión, lo que liga la plaqueta a las fibras de colágeno expuestas.
La agregación plaquetaria ocurre poco después de la adhesión. La secreción del contenido de los gránulos de plaquetas actúa como mediadora. La liberación del contenido corporal denso tiene particular importancia porque se requiere calcio para el componente de coagulación de la hemostasia y el ADP es un mediador de la agregación plaquetaria. La liberación de ADP facilita también su liberación de otras plaquetas, lo cual conduce a la amplificación del proceso de agregación. Además de ADP, las plaquetas secretan la prostaglandina TXA2, que es un estímulo importante para la agregación plaquetaria. Las acciones combinadas de ADP y TXA2 conducen a la expansión del agregado plaquetario que se convierte en el tapón hemostático primario. El tapón plaquetario se estabiliza conforme la vía de coagulación se activa en la superficie plaquetaria y el fibrinógeno se convierte en fibrina. Esto crea una red de fibrina que cimienta las plaquetas y otros componentes sanguíneos juntos (figura 26-1). La selectina P también es parte del proceso de agregación plaquetaria puesto que se une a leucocitos, que, con sustancias plaquetarias como FCDP, participan en la cicatrización de la pared del vaso.
La membrana plaquetaria desempeña una función relevante en la adhesión de plaquetas y el proceso de coagulación. La cubierta de glucoproteínas en su superficie controla las interacciones con el endotelio vascular. En condiciones normales, las plaquetas evitan la adhesión al endotelio, pero interactúan con áreas lesionadas de la pared vascular y el colágeno expuesto más profundo. Los receptores de glucoproteína (GPIIb/IIIa) en la membrana plaquetaria se unen a fibrinógeno y vinculan plaquetas. La formación defectuosa del tapón plaquetario causa hemorragia en personas con insuficiencia de plaquetas o FvW. Además de sellar las rupturas vasculares, las plaquetas desempeñan una función casi continua en el mantenimiento de la integridad vascular normal. Pueden aportarfactores de crecimiento para las células musculares lisas endoteliales y arteriales. Las personas con insuficiencia plaquetaria tienen permeabilidad capilar incrementada y sufren pequeñas hemorragias cutáneas por traumatismos o cambios en la presión arterial.
Los inhibidores de la agregación plaquetaria, incluidos ácido acetilsalicílico, clopidogrel y ticlopidina, pueden consumirse para evitar la agregación plaquetaria y la formación de coágulos en personas que están en riesgo de infarto de miocardio, accidente cerebrovascular o enfermedad arterial periférica. El tratamiento con ácido acetilsalicílico en dosis bajas inhibe la síntesis de prostaglandina, incluida TXA2.
El clopidogrel y la ticlopidina alcanzan sus efectos plaquetarios al inhibir la vía de ADP en las plaquetas. A diferencia del ácido acetilsalicílico, estos fármacos tienen un efecto en la síntesis de prostaglandina. Tanto el clopidogrel como la ticlopidina prolongan el tiempo de hemorragia. Sin embargo, los efectos secundarios más graves de la ticlopidina son neutropenia y púrpura trombocitopénica trombótica.
Estudios de ensayos aleatorizados demostraron que el clopidogrel en combinación con ácido acetilsalicílico se relaciona con una reducción de accidentes cardíacos mayores, similar a combinar ticlopidina con ácido acetilsalicílico, y parecieron más seguros. Las Antiplatelet Therapy Guidelines publicadas de manera conjunta por la American Heart Association y el American College of Cardiology se actualizaron para sugerir que el empleo del inhibidor de la bomba de protones con tratamiento antiplaquetario, en específico con clopidogrel, podría causar efectos cardíacos adversos por la interacción del fármaco con el citocromo P450. Así, aunque agregar un inhibidor de la bomba de protones a la mayor parte del tratamiento antiplaquetario parece ser eficaz, se recomienda que el clopidogrel se administre sin un inhibidor de la bomba de protones por los posibles efectos secundarios cardiovasculares negativos.
Los fármacos que actúan como inhibidores del receptor GPIIb/IIIa (tirofibán, eptifibatida, abciximab) se desarrollaron para utilizarse en el tratamiento de síndromes coronarios agudos.
Coagulación sanguínea
La cascada de coagulación es parte del proceso hemostático. Es un proceso por pasos que promueve la conversión de la proteína plasmática soluble, el fibrinógeno, en fibrina. Las hebras de fibrina insoluble crean una red que pega las plaquetas y otros componentes de la sangre para formar el coágulo.
Muchas sustancias que promueven la formación del coágulo (factores de procoagulación) o lainhiben (factores de anti-coagulación) controlan el proceso de coagulación. Cada uno de los factores de procoagulación o coagulación, identificados con números romanos, realiza un paso específico en el proceso de coagulación. La activación de un factor de procoagulación o proenzima está diseñada para activar el siguiente factor en la secuencia (efecto de cascada). Como la mayoría de los factores de procoagulación inactivos está presente en la sangre todo el tiempo, el proceso de varios pasos asegura que no tenga lugar un episodio masivo de coagulación intravascular. Esto también significa que las anomalías del proceso de coagulación se presentan cuando uno o más de los factores son insuficientes o cuando las condiciones conducen a la activación inapropiada de alguno de los pasos.
Casi todos los factores de coagulación son proteínas sintetizadas en el hígado. La vitamina K es necesaria para la síntesis de factores II, VII, IX y X, protrombina y proteína C. Si hay insuficiencia de vitamina K o insuficiencia hepática que imposibiliten la creación de suficiente protrombina, se desarrolla una tendencia hemorrágica. El calcio (factor iv) es necesario en todos excepto los 2 primeros pasos del proceso de coagulación. El cuerpo suele tener cantidades suficientes de calcio para estas reacciones. La inactivación del ion calcio evita que la sangre se coagule cuando se elimina del cuerpo. La adición de citrato a la sangre almacenada para fines de transfusión evita la coagulación al formar un quelato con el ion calcio. El ácido etilendiaminotetraacético (AEDT), otro quelante, a menudo se agrega a muestras de sangre que se utilizan para análisis en el laboratorio clínico.
El proceso de coagulación es resultado de la activación de lo que tradicionalmente se designa como las vías intrínseca y extrínseca, las cuales forman el activador de protrombina (figura 26-3).
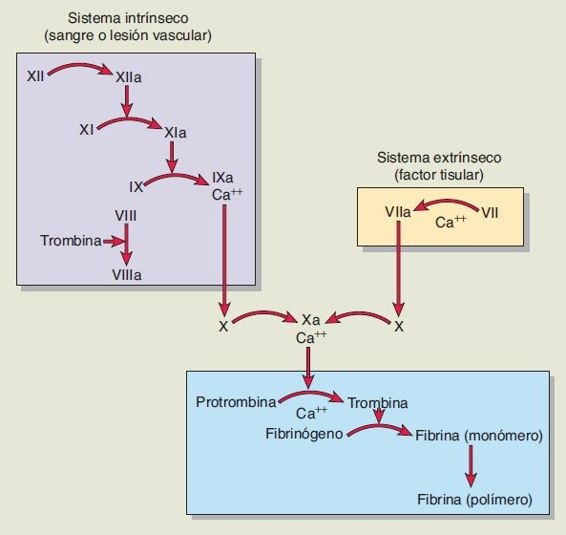
La vía intrínseca, que es un proceso hasta cierto punto lento (puede producir coagulación en 1 min a 6 min), comienza en la circulación con la activación del factor XII1.
La vía extrínseca, que es un proceso mucho más rápido (puede producir coagulación en 15 s), inicia con el traumatismo del vaso sanguíneo o los tejidos circundantes y la liberación del factor tisular o tromboplastina tisular, una lipoproteína adhesiva, de las células subendoteliales. Se compone de fosfolípidos de las membranas junto con un complejo de lipoproteína que actúa como una enzima proteolítica.
Los pasos terminales en ambas vías son los mismos: activación del factor X y conversión de protrombina en trombina. La trombina actúa luego como una enzima para convertir fibrinógeno en fibrina, el material que estabiliza un coágulo. Ambas vías son necesarias para la hemostasia normal y existen muchas interrelaciones entre ellas. Cada sistema se activa cuando la sangre sale del sistema vascular. El sistema intrínseco se activa cuando la sangre entra en contacto con el colágeno en la pared del vaso lesionado. El sistema extrínseco se activa cuando la sangre se expone a extractos tisulares. Sin embargo, la hemorragia secundaria por defectos en el sistema extrínseco no suele ser tan grave como la que resulta de defectos en la vía intrínseca.
La coagulación sanguínea está regulada por varios anti-coagulantes naturales. La antitrombina III inactiva los factores de coagulación y neutraliza la trombina, la última enzima en la vía para la conversión de fibrinógeno en fibrina. Cuando la anti-trombina III forma un complejo con la heparina de origen natural, su acción se acelera para inactivar la trombina, el factor Xa y otros factores de coagulación. Esta activación compleja confiere protección contra la formación de trombos no controlada en la superficie endotelial.
La proteína C, una proteína plasmática, actúa como un anti-coagulante al inactivar los factores V y VIII. El antígeno de proteína C, o antígeno PC (factor V Leiden), se produce en el hígado y previene la trombosis. La insuficiencia de proteína C es congénita del 35% al 58% de las veces, pero también puede ser adquirida si se tiene insuficiencia hepática grave, insuficiencia de vitamina K o neoplasia. Este trastorno es un defecto hereditario en el factor V y confiere mayor riesgo de coagulación. Puede medirse con una prueba de resistencia de proteína C y el intervalo normal debe estar entre 0,6 y 1,25 de antígeno PC normal. Las mujeres con factor V Leiden combinado con la influencia protrombótica del embarazo están en alto riesgo de tener resultados adversos de la gestación como trastornos de tromboembolia venosa, preeclampsia, pérdida del feto y desprendimiento de la placenta.
La proteína S, otra proteína plasmática, acelera la acción de la proteína C. Una insuficiencia de proteína C o S pone a los individuos en riesgo de trombosis. Se realiza una prueba de proteína S para determinar si la insuficiencia es heredada o adquirida porque a menudo las personas con trastornos autoinmunitarios están en riesgo de tener carencia de proteína S. El intervalo normal para las mujeres es de 0,5 a 1,2 de la actividad normal y para varones, de 0,6 a 1,3.
La plasmina descompone la fibrina en productos de degradación de fibrina que actúan como anticoagulantes. Se sugiere que algunos de estos anticoagulantes naturales podrían desempeñar una función en la hemorragia que se observa con coagulación intravascular diseminada (CID).
Los fármacos anticoagulantes warfarina y heparina se utilizan para prevenir trastornos tromboembólicos, como trombosis venosa profunda y embolismo pulmonar.
La warfarina disminuye la protrombina y otros factores de procoagulación. Altera la vitamina K de un modo que reduce su capacidad para participar en la síntesis de los factores de coagulación dependientes de la vitamina K en el hígado. La warfarina se absorbe fácilmente después de la administración oral. Su efecto máximo toma 36 h a 72 h debido a las diferentes semividas de los factores de coagulación preformados que permanecen en la circulación.
La heparina se forma de modo natural y es liberada en cantidades pequeñas por los mastocitos en el tejido conectivo que rodea los capilares. Las preparaciones farmacológicas de heparina se extraen de tejidos animales. La heparina se une a antitrombina III, lo que causa un cambio de conformación que incrementa la capacidad de la anti-trombina III para inactivar trombina, factor Xa y otros factores de coagulación. Al promover la inactivación de los factores de coagulación, la heparina, por último, suprime la formación de fibrina.
La heparina es incapaz de cruzar las membranas del tubo digestivo y debe administrarse mediante inyección, casi siempre por infusión intravenosa. Las heparinas de bajo peso molecular se desarrollaron para inhibir la activación del factor X, pero tienen poco efecto en la trombina y otros factores de coagulación. Las heparinas de bajo peso molecular se administran por inyección subcutánea y requieren administración y vigilancia menos frecuente que la heparina estándar (no fraccionada).
Las complicaciones potenciales del consumo de warfarina son muchas. Además, la persona necesita someterse a pruebas de laboratorio frecuentes de su tiempo de anticoagulante con una prueba de la International Normalized Ratio (INR). Un nuevo anticoagulante oral que tiene menos complicaciones y requiere menos control es el dabigatrán, que se emplea de modo gradual en personas que tienen fibrilación auricular.
Los Centers for Disease Control (2010) estimaron que en 2050 habrá 12 millones de personas con fibrilación auricular. Por lo tanto, la aparición del dabigatrán es oportuna. Además, Freeman (2010) investigó el costo de utilizar dabigatrán en lugar de warfarina y halló que el dabigatrán es más efectivo en cuanto a costo cuando se emplea en personas con fibrilación auricular.
Retracción del coágulo
En condiciones normales, el coágulo se retrae dentro de los 20 min a 60 min que siguen a su formación, lo que contribuye a la hemostasia al exprimir el suero del coágulo y a unir los bordes del vaso roto. Las plaquetas, por efecto de la acción de sus filamentos de actina y miosina, contribuyen también a la retracción del coágulo y la hemostasia. La retracción del coágulo requiere grandes cantidades de plaquetas e indica un recuento bajo de plaquetas cuando no se efectúa.
Disolución del coágulo
La disolución de un coágulo de sangre comienza poco después de su formación. Esto permite que el flujo sanguíneo se restablezca y que la reparación tisular tenga lugar. El proceso por el cual se disuelve un coágulo de sangre se llama fibrinólisis. Así como la formación del coágulo, la disolución requiere una secuencia de pasos controlados por activadores e inhibidores. Elplasminógeno, la proenzima para el proceso fibrinolítico, normalmente se presenta en la sangre en su forma inactiva. Se convierte en su forma activa, la plasmina, mediante activadores de plasminógeno formados en el endotelio vascular, el hígado y los riñones. La plasmina formada de plasminógeno digiere las hebras de fibrina del coágulo y ciertos factores de coagulación, como fibrinógeno, factor V, factor VIII, protrombina y factor XII. La plasmina circulante es inactivada con rapidez por el inhibidor de α2-plasmina, que limita el proceso fibrinolítico al coágulo local y evita que se presente en la circulación completa.
Dos activadores de plasminógeno de origen natural son el activador de plasminógeno tipo tisular y el activador de plasminógeno tipo urocinasa. El hígado, el plasma y el endotelio vascular son las principales fuentes de activadores fisiológicos. Estos activadores se liberan en respuesta a diversos estímulos, incluidos fármacos vasoactivos, oclusión venosa, temperatura corporal alta y ejercicio. Los activadores son inestables e inactivados rápidamente por inhibidores sintetizados por el endotelio y el hígado. Por esta razón, la enfermedad hepática crónica puede alterar la actividad fibrinolítica.
Concentraciones altas de un inhibidor principal, el inhibidor activador de plasminógeno 1, se han relacionado con trombosis venosa profunda, enfermedad de la arteria coronaria e infarto de miocardio. Varios activadores de plasminógeno tisular (altepasa, reteplasa, tececteplasa), producidos mediante tecnología de ADN recombinante, están disponibles para el tratamiento de infarto de miocardio agudo, accidente cerebrovascular isquémico agudo y embolismo pulmonar.