03. Trastornos hemorrágicos
Los trastornos hemorrágicos o el deterioro de la coagulación sanguínea pueden deberse a defectos en alguno de los factores que contribuyen a la hemostasia. Es posible que la hemorragia sea resultado de trastornos relacionados con el número o la función de las plaquetas, factores de coagulación e integridad de los vasos sanguíneos.
Hemorragia relacionada con trastornos plaquetarios
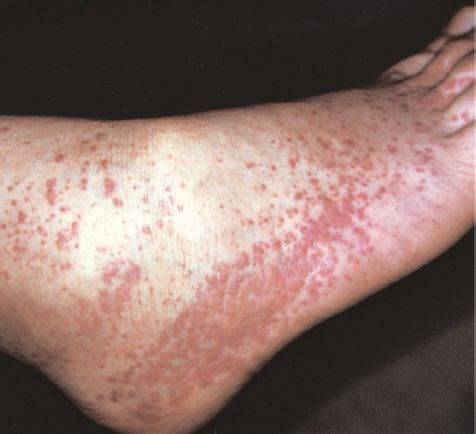
La hemorragia debida a trastornos plaquetarios refleja un disminución de la cantidad de plaquetas secundaria a la producción reducida, destrucción incrementada o función deteriorada. La hemorragia espontánea de trastornos plaquetarios con mucha frecuencia se relaciona con vasos pequeños de las membranas mucosas y la piel. Los sitios frecuentes de hemorragia son las membranas mucosas de nariz, boca, tubo digestivo y cavidad uterina. La hemorragia cutánea se ve como pequeñas hemorragias (petequias) y zonas púrpuras de magulladuras (púrpura) en áreas dependientes en las que la presión capilar es mayor (figura 26-4). Las petequias se ven casi exclusivamente en el padecimiento de insuficiencia plaquetaria y no de disfunción. La hemorragia de vasos intracraneales es un peligro raro con agotamiento plaquetario grave.
Trombocitopenia
Una reducción del número de plaquetas, también conocida como trombocitopenia, es una causa importante de hemorragia generalizada. Trombocitopenia suele referirse a disminución del número de plaquetas circulantes a un nivel menor de 150.000/μl. Entre mayor sea la disminución del recuento de plaquetas, mayor será el riesgo de hemorragia. La trombocitopenia puede ser consecuencia de descenso de la producción de plaquetas, aumento del secuestro de plaquetas en el bazo o reducción de la supervivencia de plaquetas.
La disminución de la producción de plaquetas por pérdida de la función de la médula ósea tiene lugar en la anemia aplásica. El reemplazo de médula ósea por células malignas, como se observa en la leucemia, también ocasiona una menor producción de plaquetas. La radioterapia y fármacos como los que se emplean en el tratamiento del cáncer pueden deprimir la función de la médula ósea y disminuir la producción plaquetaria. Es posible que la infección con virus de inmunodeficiencia humana (VIH) o citomegalovirus suprima la producción de megacariocitos, los precursores de plaquetas.
La producción de plaquetas puede ser normal, pero es factible que se acumulen de modo excesivo en el bazo. Aunque el bazo normalmente secuestra del 30% al 40% de las plaquetas antes de liberarlas a la circulación, la proporción puede ser de hasta el 90% cuando se agranda en la esplenomegalia. Si es necesario, la trombocitopenia hiperesplénica puede tratarse con esplenectomía.
La reducción de la supervivencia plaquetaria se debe a diversos mecanismos inmunitarios y autoinmunitarios. Anticuerpos antiplaquetarios pueden destruir plaquetas. Los anticuerpos pueden dirigirse contra los autoantígenos plaquetarios o contra antígenos en las plaquetas de transfusiones sanguíneas o embarazo. Los anticuerpos se dirigen a las glucoproteínas de la membrana plaquetaria GPIIb/IIIa y GPIb/IX. La destrucción no inmunitaria de plaquetas es consecuencia de lesión mecánica por válvulas cardíacas prostéticas o hipertensión maligna, que estrechan vasos pequeños. En CID aguda o púrpura trombocitopénica trombótica, el consumo excesivo de plaquetas da lugar a insuficiencia.
Trombocitopenia inducida por fármacos
Algunos fármacos, como quinina, quinidina y ciertos antibióticos que contienen sulfas, pueden inducir la trombocitopenia. Estos medicamentos inducen una respuesta antígeno-anticuerpo y la formación de complejos inmunes que ocasionan destrucción de plaquetas por lisis mediada por complemento. Las personas con trombocitopenia relacionada con fármacos experimentan una caída rápida del recuento plaquetario de 2 a 3 días de volver a tomar el medicamento o 7 o más días (es decir, el tiempo necesario para preparar una respuesta inmunitaria) después de empezar uno por primera vez. El recuento plaquetario aumenta con rapidez tras descontinuar el fármaco.
Trombocitopenia inducida por heparina
La trombocitopenia inducida por heparina (TIH) se relaciona con la heparina farmacológica anticoagulante. El 10% de las personas tratadas con heparinamanifiesta trombocitopenia transitoria leve en 2 a 5 días de empezar el fármaco. Sin embargo, alrededor del 1% al 5% de quienes reciben heparina sufre accidentes trombóticos que ponen en riesgo la vida 1 a 2 semanas después de iniciar el tratamiento. La TIH se debe a una reacción inmunitaria dirigida contra un complejo de heparina y factor plaquetario 4, un componente normal de los gránulos plaquetarios que se une con firmeza a la heparina. El enlace de un anticuerpo con el factor plaquetario produce complejos inmunitarios que activan las plaquetas restantes, lo que da lugar a trombosis. Además, las partículas de plaquetas protrombóticas y la inducción del factor tisular continúan promoviendo la coagulación.
El tratamiento de la TIH requiere descontinuar de inmediato el tratamiento con heparina y el consumo alternativo de anticoagulantes para evitar que la trombosis recurra. Se ha demostrado que la heparina más reciente de peso molecular bajo es efectiva para reducir la incidencia de complicaciones inducidas por heparina en comparación con la forma anterior del fármaco de alto peso molecular.
Púrpura trombocitopénica inmunitaria
La púrpura trombocitopénica inmunitaria (PTI) es una enfermedad autoinmunitaria que causa formación de anticuerpos plaquetarios y destrucción excesiva de plaquetas. La enfermedad puede tener lugar en ausencia de cualquier factor de riesgo conocido (PTI primaria o idiopática) o como un trastorno secundario debido a una alteración subyacente y como una enfermedad aguda (duración de 6 meses o menos) o crónica. Algunas formas secundarias de PTI se relacionan con el síndrome de inmunodeficiencia adquirida (sida), lupus eritematoso sistémico, síndrome antifosfolípido, leucemia linfocítica crónica, linfoma, hepatitis C y medicamentos como heparina y quinidina.
Un trastorno de PTI aguda ocurre en niños pequeños (5 años de edad) y, por lo general, sigue a una infección viral. Se caracteriza por inicio repentino de petequias y púrpura, y suele ser un trastorno autolimitado que no requiere tratamiento. La mayoría de niños se recupera en pocas semanas. En contraste, la PTI primaria a menudo es un trastorno crónico en adultos con un inicio insidioso que pocas veces sigue a una infección.
Etiología y patogénesis
Se cree que la trombocitopenia que acompaña al PTI es resultado de múltiples mecanismos que incluyen anticuerpos antiplaquetarios contra glucoproteínas (IIb/IIIa y Ib/IX) en la membrana plaquetaria. Las plaquetas, que se hacen más susceptibles a la fagocitosis por el anticuerpo, se destruyen en el bazo. Los niveles plasmáticos de trombopoyetina, el factor principal que estimula el crecimiento y desarrollo de megacariocitos, no son altos en personas con PTI. Las pruebas indican que el PTI es causado por disfunción de células T, en específico células reguladoras CD4 y T, que activan la respuesta inmunitaria y pasan a trombocitopenia.
Manifestaciones clínicas
Las manifestaciones de PTI incluyen antecedentes de equimosis, sangrado de encías, epistaxis (es decir, hemorragia nasal), melena y hemorragia menstrual anómala en quienes tienen recuento plaquetario moderadamente reducido. Ya que el bazo es el sitio donde las plaquetas se destruyen, puede observarse agrandamiento esplénico. La afección se descubre de modo incidental o como resultado de signos de hemorragia, a menudo hacia la piel (es decir, púrpura y petequias) o la mucosa bucal.
Diagnóstico y tratamiento
El diagnóstico de la PTI suele basarse en trombocitopenia grave (recuento plaquetario <20.000/μl a 30.000/μl) y exclusión de otras causas. Existen pruebas para los anticuerpos unidos a plaquetas, pero carecen de especificidad (ej. reaccionan con anticuerpos plaquetarios de otras fuentes). La forma secundaria de PTI a veces imita la forma idiopática del trastorno, por lo tanto, el diagnóstico se establece sólo después de excluir otras causas conocidas de trombocitopenia.
La decisión de tratar la PTI se basa en el recuento de plaquetas y el grado de hemorragia. Muchas personas con PTI están bien sin tratamiento. Los corticoesteroides casi siempre se utilizan como tratamiento inicial. Otros tratamientos iniciales efectivos incluyen globulina inmune intravenosa. Sin embargo, este tratamiento es costoso y el efecto beneficioso dura sólo 1 a 2 semanas.
Púrpura trombocitopénica trombótica
La púrpura trombocitopénica trombótica (PTT) es una combinación de trombocitopenia, anemia hemolítica, insuficiencia renal, fiebre y anomalías neurológicas. Es un trastorno raro que probablemente resulta de la introducción de sustancias de agregación plaquetaria a la circulación. La etiología subyacente de muchos casos es la insuficiencia de una enzima (designada ADAMTS 13, por sus siglas en inglés) que degrada multímeros de peso molecular grande de FvW, lo que permite que se acumulen y causen agregación plaquetaria y adhesión al endotelio.
Etiología y patogénesis
Es posible que la insuficiencia enzimática sea heredada o adquirida como una consecuencia de anticuerpos dirigidos contra la enzima. La PTT suele afectar a personas previamente sanas, pero también se relaciona con enfermedades del colágeno autoinmunitarias, fármacos, infecciones como VIH y embarazo. El trastorno es similar a la CID, pero no se vincula con el sistema de coagulación. Las toxinas producidas por algunas cepas de Escherichia coli (ej. E. coli O157: H7) causan lesión endotelial y producen una afección similar, el síndrome urémico hemolítico (SUH).
El inicio de la PTT es abrupto y el resultado final puede ser fatal. Las oclusiones vasculares diseminadas se deben a trombos en arteriolas y capilares de muchos órganos, incluido corazón, cerebro y riñones. Los eritrocitos se fragmentan conforme circulan por los vasos parcialmente ocluidos, lo que ocasiona anemia e ictericia.
Manifestaciones clínicas y tratamiento
Las manifestaciones clínicas comprenden púrpura, petequias, hemorragia vaginal y síntomas neurológicos que van desde cefalea hasta convulsiones y conciencia alterada. El tratamiento de urgencia para la PTT implica plasmaféresis, un procedimiento que conlleva la remoción de plasma de la sangre extraída y reemplazo con plasma recién congelado.
La infusión de plasma proporciona la enzima insuficiente. Con la plasmaféresis y el tratamiento de infusión de plasma, la recuperación es completa en 80% de los casos.
Función plaquetaria deteriorada
El trastorno de la función plaquetaria (denominado también trombocitopatía) puede deberse a enfermedades de adhesión heredadas (ej. enfermedad de von Willebrand) o defectos adquiridos ocasionados por fármacos, enfermedad o intervención quirúrgica que requiere circulación extracorpórea (es decir, derivación cardiopulmonar). La función plaquetaria insuficiente también es común en la uremia, tal vez como resultado de productos de desecho no excretados.
La administración de ácido acetilsalicílico y otros medicamentos antiinflamatorios no esteroideos (AINE) es la causa más frecuente de disfunción plaquetaria. El ácido acetilsalicílico produce acetilación irreversible de la actividad de la ciclooxigenasa plaquetaria y, en consecuencia, la síntesis de TXA2 necesaria para la agregación plaquetaria. En contraste con los efectos del ácido acetilsalicílico, la inhibición de ciclooxigenasa por otros AINE es reversible y dura sólo el tiempo que la acción del fármaco persiste. Se recurre al ácido acetilsalicílico (81 mg al día) para evitar la formación de trombos arteriales y reducir el riesgo de ataque cardíaco y accidente cerebrovascular. En el recuadro 26-2 se enumeran otros medicamentos que afectan la función plaquetaria.
Recuadro 26-2. Fármacos que pueden predisponer a hemorragia (la lista no es exhaustiva):
- Interferencia con la producción o función plaquetarias
- Acetazolamida.
- Antimetabolitos y fármacos anticáncer.
- Antibióticos, como penicilina y cefalosporinas.
- Ácido acetilsalicílico y salicilatos.
- Carbamacepina.
- Clofibrato.
- Colchicina.
- Dipiradamol.
- Diuréticos tiacídicos.
- Sales de oro.
- Heparina.
- AINE.
- Derivados de quinina (quinidina e hidroxicloroquina).
- Sulfonamidas.
- Interferencia con factores de coagulación
- Amiodarona.
- Esteroides anabólicos.
- Warfarina.
- Heparina.
- Disminución de las concentraciones de vitamina K
- Antibióticos.
- Clofibrato.
Hemorragia relacionada con insuficiencias del factor de coagulación
Los defectos de coagulación sanguínea pueden ser consecuencia de insuficiencia o función alterada de uno o más de los factores de coagulación, incluido el FvW. Las insuficiencias surgen como resultado de enfermedad heredada o síntesis defectuosa o incremento del consumo de factores de coagulación. La hemorragia secundaria a las insuficiencias del factor de coagulación suelen tener lugar después de lesión o traumatismo. Son comunes las magulladuras grandes, hematomas y hemorragia prolongada hacia el tracto gastrointestinal, el urinario o las articulaciones.
Trastornos heredados
La enfermedad de von Willebrand y la hemofilia (A y B) son 2 de los trastornos hemorrágicos heredados más frecuentes. La enfermedad de von Willebrand se considera la coagulopatía here-dada más común y afecta a cerca del 1% al 2% de la población. La hemofilia A (insuficiencia del factor VIII ) afecta a 1 de 5.000 nacimientos vivos de varones. La hemofilia B (insuficiencia del factor ix) ocurre en alrededor de 1 de 20.000 personas, lo que explica 15% de personas con hemofilia. Es genética y clínicamente similar a la hemofilia A. La enfermedad de von Willebrand y la hemofilia A se deben a defectos relacionados con el complejo del factor VIII-FvW. El FvW, que es sintetizado por el endotelio y los megacariociotos, se requiere para la adhesión de plaquetas a la matriz subendotelial del vaso sanguíneo. Sirve también como portador del factor VIII y es importante para la estabilidad del factor VIII en la circulación al prevenir su proteólisis. El hígado y las células endoteliales producen la proteína coagulante de la porción funcional del factor VIII. Así, los factores VIII y FvW, que se sintetizan por separado, entran juntos al plasma y circulan en él como una unidad que promueve la coagulación y la adhesión plaquetaria a la pared del vaso.
Enfermedad de von Willebrand
La enfermedad de von Willebrand es un trastorno hemorrágico hereditario frecuente que se caracteriza por una insuficiencia o un defecto en FvW. Se han descrito hasta 20 variantes de la enfermedad de von Willebrand. Estas variantes pueden agruparse en 2 categorías: tipos 1 y 3, que se relacionan con bajos niveles de FvW y tipo 2, que se distingue por defectos en el FvW.
Clasificación
El tipo 1, un trastorno autosómico dominante, constituye cerca de 70% de los casos y es relativamente leve. El tipo 2, también un trastorno autosómico dominante, explica cerca del 25% de los casos y se relaciona con hemorragia leve a moderada. El tipo 3, que es un trastorno autosómico recesivo más o menos raro, se relaciona con niveles en extremo bajos de FvW funcional y, por consiguiente, manifestaciones clínicas graves. Las personas con enfermedad de von Willebrand tienen un defecto compuesto que compromete la función plaquetaria y la vía de la coagulación.
Manifestaciones clínicas
Incluyen hemorragia espontánea nasal, bucal y del tubo digestivo, flujo menstrual excesivo y tiempo de hemorragia prolongado en presencia de un recuento plaquetario normal. Casi todos los casos (es decir, los tipos 1 y 2) son leves y no requieren tratamiento, y muchas personas con el trastorno se diagnostican cuando una intervención quirúrgica o extracción dental produce hemorragia prolongada. En casos graves (es decir, tipo 3), la hemorragia gastrointestinal que pone en riesgo la vida y la hemorragia de las articulaciones pueden ser similares a la que se observa en la hemofilia. La hemorragia relacionada con la enfermedad de von Willebrand suele ser leve y no se administra ningún tratamiento sistemático distinto a la evasión del ácido acetilsalicílico.
Hemofilia A
La hemofilia A es un trastorno recesivo ligado a X que afecta sobre todo a los varones.Aunque es hereditario, no hay antecedentes familiares del trastorno en casi el 30% de los casos recién diagnosticados, lo que sugiere que surgió como una nueva mutación en el gen del factor VIII4.
Alrededor del 90% de las personas con hemofilia produce cantidades insuficientes del factor y 10% produce una forma defectuosa. El porcentaje de actividad normal del factor VIII en la circulación depende del defecto genético y determina la gravedad de la hemofilia (es decir, del 6% al 30% en la hemofilia leve, del 2% al 5% en la hemofilia moderada y el 1% o menos en formas graves de hemofilia). En las formas leve o moderada de la enfermedad, la hemorragia no suele presentarse a menos que haya una lesión local o traumatismo como intervención quirúrgica o procedimiento dental. Es posible que el trastorno leve no se detecte en la infancia. En la hemofilia grave, la hemorragia casi siempre ocurre en la infancia (ej. puede advertirse en el momento de la circuncisión), es espontánea, grave y a menudo se presenta varias veces al mes.
Manifestaciones clínicas
De manera característica, la hemorragia tiene lugar en tejidos blandos, tubo digestivo y cadera, rodilla, codo y articulaciones del tobillo. La hemorragia espontánea de las articulaciones suele iniciar cuando un niño comienza a caminar. Con frecuencia, la articulación objetivo es propensa a hemorragia repetida. La hemorragia causa inflamación de la membrana sinovial, con dolor agudo e hinchazón. Sin el tratamiento adecuado, la hemorragia crónica y la inflamación producen fibrosis articular y contracturas, cuyo resultado es incapacidad mayor.
Tratamiento
La prevención del traumatismo es importante en personas con hemofilia. Debe evitarse el ácido acetilsalicílico y otros AINE que afectan la función plaquetaria. El tratamiento de restitución del factor VIII que se administra en casa reduce el daño musculoesquelético característico. Se inicia cuando la hemorragia se presenta o como profilaxis con episodios hemorrágicos repetidos. Es posible que los productos recombinantes y las bombas de infusión continua sean preventivos más que terapéuticos. El desarrollo de anticuerpos inhibidores para el factor recombinante VIII aún es una complicación importante del tratamiento.
La clonación del gen del factor VIII y el avance en los sistemas de creación de genes son esperanzadores con respecto a que la hemofilia A pueda curarse con el tratamiento de restitución génica. La detección del portador y el diagnóstico parental pueden realizarse ahora mediante análisis de mutación directa de genes o estudios de enlace de ADN. La amniocentesis prenatal o muestreo de vellosidades coriónicas se utiliza para predecir complicaciones y determinar el tratamiento. Por último, puede emplearse para seleccionar a pacientes para la adición de genes.
Trastornos adquiridos
Los factores de coagulación V, VII, IX, X, XI y XII, la protrombina y el fibrinógeno se sintetizan en el hígado. En la enfermedad hepática, la síntesis de factores de coagulación se reduce, lo que podría producir hemorragia. De los factores de coagulación que se sintetizan en el hígado, los factores II, VII, IX y, X, y la protrombina requieren la presencia de vitamina K para su actividad normal. En la insuficiencia de vitamina K, el hígado produce el factor de coagulación, pero en una forma inactiva.
La vitamina K es una vitamina liposoluble que bacterias intestinales sintetizan de manera continua. Esto significa que la insuficiencia de vitamina K es improbable a menos que la síntesis intestinal se interrumpa o la absorción de la vitamina se afecte. La insuficiencia de vitamina K puede presentarse en el neonato antes que la flora intestinal se establezca. También puede ocurrir como resultado del tratamiento con antibióticos de amplio espectro que destruyen la flora intestinal. Como la vitamina K es liposoluble, su absorción requiere sales biliares. Es posible que la insuficiencia de vitamina K sea consecuencia de absorción alterada de grasa secundaria a la enfermedad hepática o de la vesícula biliar.
Hemorragia relacionada con trastornos vasculares
La hemorragia por trastornos vasculares a veces se denomina púrpura no trombocitopénica. Estos trastornos pueden ser el resultado de paredes vasculares estructuralmente débiles o de daño de los vasos por inflamación o respuestas inmunitarias. Muy a menudo se caracterizan por equimosis fácil, aparición espontánea de petequias y púrpura en piel, y membranas mucosas. El recuento plaquetario y los resultados de otras pruebas de factores de coagulación son normales en personas con trastornos hemorrágicos causados por defectos vasculares.
Entre los trastornos vasculares que causan hemorragia están la telangiectasia hemorrágica, un trastorno autosómico frecuente caracterizado por capilares dilatados de pared delgada y arteriolas; insuficiencia de vitamina C (es decir, escorbuto), que conduce a síntesis insuficiente de colágeno y fallo de las células endoteliales para cementarse juntas de forma adecuada, lo que hace que la pared vascular sea frágil; enfermedad de Cushing, que causa atrofia proteínica y pérdida de soporte del tejido vascular por exceso de cortisol; y púrpura senil (es decir, equimosis en adultos mayores), ocasionada por deterioro de la síntesis de colágeno en el proceso de envejecimiento. Los defectos vasculares también tienen lugar en el curso de la CID o como consecuencia de microtrombos y tratamiento con costicoesteroides.
Coagulación intravascular diseminada
La CID es una paradoja en la secuencia hemostática y se caracteriza por coagulación generalizada y hemorragia en el compartimiento vascular. No es una enfermedad primaria pero se observa como una complicación de una amplia variedad de afecciones. La CID comienza con activación masiva de la secuencia de coagulación por la generación no regulada de trombina, que conduce a la formación sistémica de fibrina. Además, los niveles de todos los anticoagulantes principales se reducen (figura 26-5).
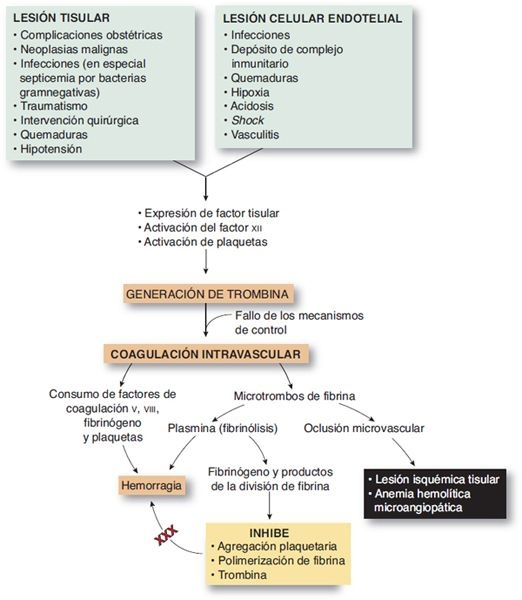
Los microtrombos resultantes causan oclusión vascular e isquemia tisular. La insuficiencia orgánica múltiple es posible. La formación del coágulo consume todas las proteínas de coagulación y plaquetas, lo que produce hemorragia grave.
Etiología y patogénesis
El trastorno puede iniciarse por la activación de la vía intrínseca, de la extrínseca o de ambas. La activación por la vía extrínseca tiene lugar con liberación de factores tisulares y se relaciona con complicaciones obstétricas, traumatismo, septicemia bacteriana y cánceres.
La vía intrínseca se puede activar mediante daño endotelial extenso, con activación del factor XII. La lesión endotelial puede ser causada por virus, infecciones, mecanismos inmunitarios, estasis de la sangre o extremos de temperatura. Las vías de anti-coagulación afectadas se vinculan también con niveles reducidos de antitrombina y el sistema anticoagulante de proteína C en la CID.
Hay evidencias de que la causa subyacente de la CID es la infección o inflamación y las citocinas (factor de necrosis tumoral, interleucina-1 y otros) que se libera en el proceso son los mediadores desencadenantes. Estas citocinas no sólo median la inflamación, sino también pueden incrementar la expresión del factor tisular en las células endoteliales y de modo simultáneo disminuir la expresión de trombomodulina. La trombomodulina, una glucoproteína presente en la membrana celular de las células endoteliales, se une a trombina y actúa como un mecanismo regulador adicional en la coagulación. Los padecimientos clínicos comunes que pueden causar CID incluyen trastornosobstétricos, que explican el 50% de los casos, traumatismo masivo, shock, septicemia y enfermedad maligna. En el recuadro 26-3 se resumen las afecciones relacionadas con CID.
Los factores que participan en los padecimientos que causan CID a menudo están interrelacionados. En complicaciones obstétricas, los factores tisulares liberados de líquido placentario necrótico o líquido tisular fetal o amniótico podrían desencadenar la CID. La hipoxia, el estado de shock y la acidosis que pueden coexistir contribuyen también al causar lesión endotelial.
Las infecciones por bacterias gramnegativas dan como resultado liberación de endotoxinas, que activan la vía extrínseca por liberación del factor tisular y la vía intrínseca por daño endotelial. Las endotoxinas inhiben también la actividad de la proteína C.
Recuadro 26-3. Padecimientos que se han relacionado con CID:
- Padecimientos obstétricos
- Desprendimiento de placenta.
- Síndrome de feto muerto.
- Preeclampsia y eclampsia.
- Embolia del líquido amniótico.
- Cánceres
- Cáncer metastático.
- Leucemia.
- Infecciones
- Infecciones bacterianas agudas (ej. meningitis meningocócica).
- Infecciones virales agudas.
- Infecciones por rickettsias (ej. fiebre manchada de las Montañas Rocosas).
- Infecciones parasitarias (ej. paludismo).
- Shock
- Shock séptico.
- Shock hipovolémico grave.
- Traumatismo e intervención quirúrgica
- Quemaduras.
- Traumatismo masivo.
- Circulación extracorpórea relacionada con intervención quirúrgica.
- Mordedura de serpiente.
- Golpe de calor.
- Padecimientos hemorrágicos
- Reacciones a transfusión sanguínea.
Manifestaciones clínicas
Aunque la coagulación y la formación de microémbolos caracterizan la CID, por lo general, susmanifestaciones agudas se relacionan de manera más directa con los problemas hemorrágicos que la acompañan. La hemorragia puede evidenciarse como petequias, púrpura, rezuma de sitios de punción o hemorragia grave.
La hemorragia posparto no controlada puede indicar CID. Es posible que los microémbolos obstruyan vasos sanguíneos y causen hipoxia tisular y daño necrótico a estructuras orgánicas, como riñones, corazón, pulmones y cerebro. En consecuencia, los signos clínicos comunes pueden deberse a insuficiencia renal, circulatoria o respiratoria, úlceras hemorrágicas agudas o convulsiones y coma. Una forma de anemia hemolítica puede presentarse cuando los eritrocitos se lesionan al pasar por vasos parcialmente bloqueados por trombos.
Tratamiento
El tratamiento de la CID se dirige a controlar la enfermedad primaria, restituir componentes de la coagulación y evitar la activación posterior de mecanismos de coagulación. Las transfusiones de plasma congelado fresco, plaquetas o crioprecipitado que contiene fibrinógeno podrían corregir la insuficiencia del factor de coagulación.