04. Equilibrio del calcio, fósforo y magnesio
Mecanismos que regulan el equilibrio del calcio, fósforo y magnesio
Calcio, fósforo y magnesio son los principales cationes del cuerpo. Se ingieren con la dieta, son absorbidos del intestino, son filtrados en el glomérulo del riñón, reabsorbidos en los túbulos renales y eliminados por la orina. Alrededor del 99% de calcio, un 85% de fosforo y del 50% al 60% de magnesio están en los huesos. La mayor parte del calcio restante (cerca del 1%), fósforo (casi el 14%) y magnesio (del 40% al 50%) está dentro de las células. Sólo una pequeña cantidad de estos 3 iones se encuentra en el LEC. Esta cantidad, pequeña pero vital, de calcio, fósforo y magnesio del LEC está directa o indirectamente regulada por la vitamina D y la hormona paratiroidea (HPT). Se cree que la calcitonina, una hormona producida por las células C en la tiroides, actúa en los riñones y hueso para eliminar el calcio de la circulación extracelular.
Vitamina D
Si bien está clasificada como vitamina, la vitamina D funciona como una hormona. Su función es conservar niveles normales de calcio y fósforo en el plasma, lo que consigue al aumentar la absorción del intestino; también es necesaria para que los huesos se formen normalmente. La vitamina D se sintetiza mediante radiación ultravioleta de 7-deshidrocolesterol, que está presente en la piel o se obtiene de los alimentos de la dieta; muchos de éstos se encuentran enriquecidos con vitamina D. Las formas sintetizadas o ingeridas de vitamina D son en esencia prohormonas que carecen de actividad biológica y tienen que pasar por una transformación metabólica para tener potencia. Una vez que la vitamina D entra a la circulación desde la piel o el intestino, se concentra en el hígado. Ahí es hidroxilada y se forma 25-hidroxivitamina D [25-(OH) D 3 ], que también se llama calcidiol. Luego es trasportada a los riñones, en donde se transforma en 1,25-(OH)2D3.
La acción principal de la forma activada de vitamina D, también conocida por calcitriol, consiste en aumentar la absorción de calcio del intestino. El calcitriol también sensibiliza al hueso para las acciones de resorción de HPT. Hay evidencias de que la vitamina D controla el crecimiento de la glándula paratiroides y de que inhibe la síntesis y la secreción de HPT. La formación de 1,25-(OH)2D3 en los riñones está regulada mediante retroalimentación por las concentraciones de calcio y fosfato que hay en el plasma. Los niveles bajos de calcio hacen que aumente HPT, la que luego incrementa la activación de la vitamina D. Una disminución del fosfato en plasma también eleva la activación de la vitamina D. Otro control de la activación renal de la vitamina D es un ciclo de retroalimentación negativo que monitorea los niveles de 1,25-(OH)2D3.
Hormona paratiroidea
La HPT, un regulador primordial del calcio y fosfato plasmáticos, es secretada por las glándulas paratiroides. Hay 4 glándulas paratiroides que están sobre la superficie dorsal de la glándula tiroides.
El regulador dominante de HPT es la concentración de calcio en el plasma. Un receptor exclusivo de calcio en la membrana celular de la paratiroides (receptor extracelular sensible al calcio) responde rápidamente a los cambios en el nivel de calcio plasmático. Cuando la concentración de calcio plasmático es alta, HPT es inhibida y el calcio se deposita en los huesos. Cuando la concentración es baja, la secreción de HPT aumenta y el calcio es movilizado desde los huesos. La respuesta a una disminución de calcio plasmático es casi inmediata, sólo tarda unos segundos. El fósforo no afecta de manera directa la secreción de HPT, sino que actúa de modo indirecto, mediante la formación de un complejo con el calcio, con lo que se reduce su concentración en el plasma.
La secreción, síntesis y acción de HPT también están influenciadas por el magnesio. Éste sirve como cofactor en la generación de energía celular y es importante en la función de sistemas de segundos mensajeros. Se cree que los efectos del magnesio en la síntesis y liberación de HPT están mediados a través de estos mecanismos. Debido a su función en la regulación de la liberación de HPT, una hipomagnesemia grave y prolongada inhibe en forma considerable las concentraciones de HPT.
La principal función de HPT es conservar la concentración de calcio del LEC. La realiza al propiciar la liberación de calcio desde el hueso, al incrementar la activación de vitamina D como un recurso para intensificar la absorción intestinal de calcio y al estimular al riñón a que conserve el calcio, a la vez que incrementa la excreción de fosfato (figura 39-11).
La HPT actúa en el hueso para acelerar la movilización y transferencia de calcio al LEC. La respuesta esquelética a la HPT es un proceso de 2 pasos. Hay una respuesta inmediata en la que el calcio que está presente en el líquido del hueso es liberado en el LEC, y una segunda respuesta que se genera con más lentitud, en la que el hueso, completamente mineralizado, es reabsorbido, lo que deriva en la liberación tanto de calcio como de fósforo.
Las acciones de HPT relacionadas con reabsorción requieren concentraciones normales, tanto de vitamina D como de magnesio. La presencia de HPT incrementa la activación de vitamina D por parte del riñón; al ser activada la vitamina D, la HPT aumenta la absorción intestinal de calcio y fósforo, así como la acción sobre el riñón para incrementar la reabsorción tubular de calcio y magnesio, a la vez que se incrementa la eliminación de fósforo. El aumento en la eliminación de fósforo garantiza que la liberación de éste desde el hueso no origine hiperfosfatemia ni aumente el riesgo de depósito de cristales de fosfato de calcio en el tejido liso.
Hipoparatiroidismo
Este trastorno refleja secreción insuficiente de HPT, lo que da como resultado hipocalcemia. La insuficiencia de HPT puede ser causada por carencia congénita de todas las glándulas paratiroideas, como sucede en el síndrome de DiGeorge. Puede haber una insuficiencia adquirida de HPT después de una intervención quirúrgica en el cuello, sobre todo si la operación se realiza para extirpar un adenoma paratiroideo, o si se trata de una tiroidectomía, o bien de una resección bilateral del cuello por cáncer.
Una forma momentánea de insuficiencia de HPT, que se presenta durante 1 o 2 días y que puede durar hasta 5 días, podría presentarse después de una intervención quirúrgica en la tiroides para inhibir la glándula paratiroides. El hipoparatiroidismo podría tener un origen autoinmune. Los anticuerpos antiparatiroideos han sido detectados en algunas personas con hipoparatiroidismo, en particular en aquellas con trastornos múltiples autoinmunes, como la diabetes tipo 1, enfermedad de Graves, enfermedad de Hashimoto y vitiligo (destrucción autoinmune de melanocitos que origina la formación de zonas totalmente blancas de la piel).
Otras causas de hipoparatiroidismo son daños por metales pesados, como el que existe en la enfermedad de Wilson, tumores metastásicos y procedimientos quirúrgicos. El deterioro funcional de la función paratiroidea se presenta con la insuficiencia de magnesio. La corrección de hipomagnesemia lleva a la desaparición rápida del trastorno.
Las manifestaciones de hipoparatiroidismo agudo, resultado de una disminución de calcio plasmático son, entre otras, tetania con calambres musculares, espasmo carpopedal y convulsiones.
Las parestesias, como hormigueo en la zona peribucal y de las manos y pies, están casi siempre presentes. La concentración baja de calcio podría causar prolongación del intervalo QT, resistencia al digital, hipotensión e insuficiencia cardíaca refractaria. Entre los síntomas de insuficiencia crónica de HPT están letargo, ansiedad y cambios de personalidad. Podría haber visión borrosa debido a cataratas que se desarrollan a lo largo de varios años. Podría haber signos extrapiramidales, como los que se observan en la enfermedad de Parkinson, dada la calcificación de los ganglios basales. El tratamiento exitoso de la hipocalcemia podría mejorar el trastorno y, a veces, en las radiografías, se relaciona con una reducción de la calcificación de los ganglios basales. Los dientes podrían estar defectuosos si el trastorno tuvo lugar durante la infancia.
El diagnóstico de hipoparatiroidismo se basa en los niveles bajos de calcio en el plasma, elevadas concentraciones de fosfato plasmático y bajos niveles de HPT plasmática. Por lo general, los niveles de magnesio en plasma se miden para verificar si hay hipomagnesemia y determinar si es ésta la causa del trastorno. La tetania hipoparatiroidea aguda se trata con gluconato de calcio por vía intravenosa, seguido de administración por vía oral de sales de calcio y vitamina D. Los complementos de magnesio se utilizan cuando el trastorno es causado por insuficiencia de magnesio.
Las personas con hipoparatiroidismo crónico se tratan con calcio por vía oral y vitamina D. Los niveles de calcio plasmático se monitorean a intervalos regulares (por lo menos cada 3 meses) para mantener el calcio plasmático dentro de unos valores ligeramente bajos pero sin síntomas. Conservar el calcio plasmático dentro de estos valores ayuda a evitar hipercalciuria y daño renal.
El seudohipoparatiroidismo es una enfermedad familiar rara que se caracteriza por resistencia específica del tejido a la HPT. Sus características son hipocalcemia, aumento de la función paratiroidea y una variedad de defectos congénitos en el crecimiento y desarrollo del esqueleto, como corta estatura y huesos metacarpianos y metatarsianos cortos. Hay variantes en la enfermedad, como las que se dan en personas que padecen seudohipoparatiroidismo con defectos congénitos y aquellas que presentan defectos congénitos y concentraciones normales de calcio y fosfato. Las manifestaciones del trastorno se deben, sobre todo, a hipocalcemia crónica. El tratamiento es similar al del hipoparatiroidismo.
Hiperparatiroidismo
Es causado por hipersecreción de HPT. Se manifiesta como un trastorno primario causado por hiperplasia (15%), un adenoma (85%) y rara vez carcinoma de las glándulas paratiroides, o como un trastorno secundario que se observa en personas con insuficiencia renal crónica o malabsorción crónica de calcio. Los adenomas paratiroideos y la hiperplasia se presentan en diversas enfermedades familiares distintas (como neoplasia endocrina múltiple [NEM] tipos 1 y 2a).
El hiperparatiroidismo primario se detecta con más frecuencia después de los 50 años de edad y es más común en mujeres que en varones. El hiperparatiroidismo causa hipercalcemia y un aumento de calcio en el filtrado de la orina, lo que da como resultado hipercalciuria y la potencial formación de cálculos renales. La reabsorción ósea crónica podría producir desmineralización difusa, fracturas patológicas y lesiones óseas quísticas. Un estudio de los huesos mediante absorciometría con rayos X de doble energía (AXDE) podría practicarse para evaluar la densidad mineral ósea (DMO). Los signos y síntomas del trastorno se relacionan con anomalías del esqueleto, exposición de los riñones a altos niveles de calcio y concentraciones elevadas de calcio plasmático.
En la actualidad, la mayoría de las personas con hiperparatiroidismo primario manifiestan un trastorno asintomático que es descubierto en el curso de pruebas químicas obligatorias.
Para diferenciar entre las 2 causas más comunes de hipercalcemia se utilizan procedimientos diagnósticos que abarcan concentraciones de calcio plasmático y niveles intactos de HPT: hiperparatiroidismo primario e hipercalcemia tumoral (HT).
En los estudios de HPT intacta se utilizan 2 anticuerpos que se unen a lugares diferentes de la HPT y están diseñados para medir de manera específica la hormona intacta, biológicamente activa. En el hiperparatiroidismo primario, los niveles de HPT intacta están elevados en el 75% al 90% de las personas afectadas, o son «normales» y hay hipercalcemia, cuando deberían estar inhibidos. En HT, los niveles de HPT intacta están inhibidos. Los estudios de imagen de la zona paratiroidea se podrían utilizar para identificar un adenoma paratiroideo. Sin embargo, la función de los estudios mediante imágenes antes de la operación y después de ella es tema de controversia. Por lo general, la operación paratiroidea es el tratamiento de elección.
El hiperparatiroidismo secundario se relaciona con hiperplasia de las glándulas paratiroides y se presenta en personas con insuficiencia renal. Cuando apenas inicia la insuficiencia, un aumento de HPT es resultado de niveles bajos de calcio plasmático y vitamina D. A medida que avanza la enfermedad, hay una reducción de receptores de vitamina D y de calcio, lo que hace que las glándulas paratiroides sean más resistentes a la regulación por retroalimentación por nivel de calcio plasmático y vitamina D. En este punto, las concentraciones altas de fosfato plasmático inducen hiperplasia de las glándulas paratiroides, independientemente del calcio y vitamina D activada.
La enfermedad ósea que se detecta en personas con hiperparatiroidismo secundario a causa de insuficiencia renal se conoce como trastorno mineral óseo enfermedad renal crónica (TMO-ERC). Esta enfermedad tiene 3 manifestaciones fisiopatológicas principales, incluido el metabolismo anómalo de calcio, fosfato, vitamina D, o HPT; calcificación de tejido liso o vasos, y anomalías en el recambio óseo. La TMO- ERC se llamó antes osteodistrofia renal. La evidencia apunta a que con TMO-ERC, los niveles altos de fósforo causaban ateroesclerosis porque aumentaban el espesor de la media-íntima de la carótida.
El tratamiento de hiperparatiroidismo incluye resolver la hipercalcemia con consumo alto de líquidos. A las personas con enfermedad leve se les recomienda mantenerse activas y beber suficientes líquidos, además de evitar antiácidos que contienen calcio, vitamina D y diuréticos con tiacida, que aumentan la reabsorción de calcio en el riñón. Se podría recomendar paratiroidectomía en las personas con hiperparatiroidismo sintomático, cálculos renales o enfermedad ósea. Evitar la hiperfosfatemia podría reducir los problemas de la TMO-ERC causada por hiperparatiroidismo secundario en insuficiencia renal. El acetato de calcio o agente sin calcio (HCl) se administran con las comidas para unir fosfato. El calcitriol, la forma activada de la vitamina D, se podría utilizar para controlar la hiperplasia de la paratiroides e inhibir la síntesis y secreción de HPT. No obstante, debido a su potente efecto en la absorción intestinal y movilización ósea, el calcitriol causa hipercalcemia. Se están perfeccionando análogos más recientes de vitamina D activada que conservan la capacidad de inhibir la función paratiroidea mientras actúan mínimamente en la reabsorción de calcio o fósforo.
Trastornos del equilibrio del calcio
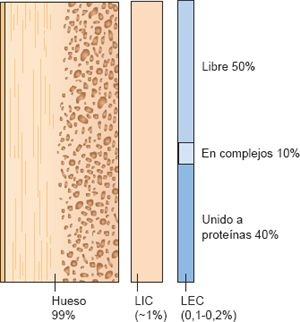
El calcio entra el cuerpo a través del tubo digestivo, es absorbido desde el intestino mediante la influencia de la vitamina D, almacenado en huesos y excretado por el riñón. Alrededor del 99% del calcio corporal está en los huesos, a los que proporciona fuerza y estabilidad para el sistema esquelético, y funciona como una fuente intercambiable para mantener los niveles de calcio extracelular. La mayor parte del calcio restante (casi el 1%) está dentro de las células y sólo entre el 0,1% y el 0,2% (cerca de 8,5 mg/dl a 10,5 mg/dl [2,1 mmol/l a 2,6 mmol/l]) del calcio restante está en el LEC.
El calcio del LEC existe en 3 formas:
- enlazado a proteínas,
- formando complejos, y
- ionizado (figura 39-11).
Alrededor del 40% del calcio del LEC está unido a proteínas plasmáticas, casi siempre albúmina, y no se difunde ni atraviesa la pared capilar para abandonar el compartimento vascular. Otro 10% forma complejos (es decir, está quelado) con sustancias como citrato, fosfato y sulfato. Esta forma no está ionizada. El 50% restante del calcio del LEC está en forma ionizada, la que es libre de dejar el compartimento vascular y participar en las funciones celulares. La concentración de calcio plasmático total fluctúa con los cambios de albúmina en plasma y de pH. El calcio ionizado participa en varias funciones. Interviene en muchas reacciones enzimáticas; produce un importante efecto en los potenciales de membrana y excitabilidad neuronal; es necesario para la contracción en el músculo del esqueleto, cardíaco y liso; participa en la liberación de hormonas, neurotransmisores y otros mensajeros químicos; determina la contractibilidad cardíaca y automaticidad a través de los canales del calcio lentos, y es esencial para la coagulación de la sangre.
La utilización de fármacos bloqueadores de los canales del calcio en los trastornos circulatorios demuestra la importancia de los iones de Ca++ en el funcionamiento normal del corazón y vasos sanguíneos. El calcio es requerido en todos, salvo en los primeros 2 pasos de la ruta intrínseca de la coagulación de la sangre. Dada su capacidad para unirse al calcio, a menudo el citrato se utiliza para evitar que se coagule la sangre que se usará en transfusiones. Ciertos estudios señalan que las concentraciones de calcio ionizado (Cai) en personas que ingresan al hospital críticamente enfermas debido a traumatismo son factores que predicen la necesidad de transfusiones múltiples; además, niveles de Cai bajos pronostican mortalidad.
Ganancias y pérdidas
Las fuentes principales de calcio en la dieta son leche y productos lácteos. Sólo del 30% al 50% del calcio de la dieta es absorbido del duodeno y yeyuno superior, el resto es eliminado con las heces. Desde la sangre, entra al intestino alrededor de 150 mg/día de calcio. La absorción neta de calcio es igual a la cantidad que es absorbida desde el intestino, menos la cantidad que se desplaza en él. El equilibrio del calcio se vuelve negativo cuando la ingesta de calcio en la dieta (y absorción de calcio) es menor que la secreción intestinal.
El calcio se deposita en los huesos y se excreta por medio de los riñones. Cerca del 60% al 65% del calcio filtrado es reabsorbido de manera pasiva en el túbulo proximal, impulsado por la reabsorción de NaCl; del 15% al 20% es reabsorbido en la rama ascendente gruesa del asa de Henle, accionado por el sistema de cotransporte de Na+/K+/2Cl+ y de 5% a 10% es reabsorbido en el túbulo contorneado distal. Este túbulo es un importante sitio regulador que controla la cantidad de calcio que entra a la orina. La HPT y quizá la vitamina D estimulan la reabsorción de calcio en este segmento de la nefrona. Los diuréticos con tiacida, que actúan en el túbulo contorneado distal, intensifican la reabsorción de calcio. Otros factores que a veces influyen en la reabsorción de calcio en esta área son las concentraciones de fosfato, glucosa e insulina.
Hipocalcemia
Representa una concentración de calcio plasmático de menos de 8,5 mg/dl (2,1 mmol/l). Se presenta en muchas formas de enfermedad grave y llega a afectar al 70% de los pacientes en las unidades de cuidados intensivos.
Causas
Las causas de hipocalcemia se dividen en 4 categorías:
- incapacidad para mover el calcio desde los depósitos de los huesos;
- pérdidas anómalas de calcio desde el riñón;
- mayor enlace con proteínas o quelación, de tal modo que mayores proporciones de calcio están en la forma no ionizada, y
- secuestro en tejido liso (tabla 39-10). Una seudohipocalcemia es causada por hipoalbuminemia. En este caso, una persona desnutrida podría tener un nivel de calcio total sérico bajo, pero sin síntomas.
El calcio plasmático está en equilibrio dinámico con el calcio de los huesos. La capacidad para mover el calcio desde el hueso depende de concentraciones correctas de HPT. Los niveles bajos de HPT puede ser resultado de formas primarias o secundarias de hipoparatiroidismo. La inhibición de liberación de HPT también podría presentarse cuando la concentración de vitamina D es elevada. Se puede utilizar la forma activada de vitamina D (calcitriol) para inhibir el hiperparatiroidismo secundario que se presenta en personas con renopatía crónica. La insuficiencia de magnesio inhibe la liberación de HPT y daña su acción en la reabsorción de hueso. Esta forma de hipocalcemia es difícil de tratar sólo con complementos de calcio, por lo que requiere corrección de la insuficiencia de magnesio.
Existe una relación inversa entre la excreción de calcio y fosfato en los riñones. La eliminación de fosfato está deteriorada en la renopatía crónica, lo que hace que disminuyan las concentraciones de calcio plasmático. La hipocalcemia e hiperfosfatemia surgen cuando el índice de filtración glomerular desciende a menos de 59 ml/min (los valores normales, los que se relacionan con el sexo, edad y complexión corporal son de cerca de 120 ml/min en mujeres jóvenes y 130 ml/min envarones jóvenes).
Sólo la forma ionizada de calcio es capaz de salir de los capilares y participar en las funciones corporales. Un cambio de pH modifica la proporción de calcio que hay en las formas enlazadas y ionizadas. Un pH ácido disminuye el enlace de calcio con las proteínas, lo que ocasiona un aumento desproporcionado de calcio ionizado, en tanto que el calcio total en el plasma permanece sin cambios. Un pH alcalino tiene el efecto contrario. Por ejemplo, la hiperventilación suficiente para causar alcalosis respiratoria origina tetania, debido a que aumenta el enlace de proteínas y calcio. Los ácidos grasos libres también incrementan la unión de calcio con albúmina, lo que origina un decremento en el calcio ionizado.
A veces se presentan aumentos de ácidos grasos libres suficientes para modificar el enlace del calcio durante situaciones estresantes que hacen que suban los niveles de adrenalina, glucagón, hormona del crecimiento y hormona adrenocorticotrópica. La heparina, fármacos β-adrenérgicos (ej. adrenalina, isoproterenol y noradrenalina) y el alcohol también aumentan los niveles de ácidos grasos libres en forma suficiente para incrementar el enlace del calcio.
El citrato, que forma complejos con calcio, a menudo se utiliza como anticoagulante en transfusiones de sangre. En teoría, el exceso de citrato en la sangre de un donador podría combinarse con el calcio de la sangre de quien la va a recibir, lo que produciría una caída considerable de calcio ionizado. Por lo regular, esta situación no se presenta, porque el hígado elimina el citrato en cuestión de minutos. Cuando se administran transfusiones de sangre a una velocidad baja, hay poco peligro de hipocalcemia causada por la unión del citrato.
La hipocalcemia es un hallazgo común en personas con pancreatitis aguda. La inflamación del páncreas ocasiona que se liberen enzimas proteolíticas y lipolíticas. Se cree que el Ca 2 + se combina con los ácidos grasos libres liberados por la lipólisis en el páncreas, lo que forma anomalías y retira el calcio de la circulación.
La insuficiencia de calcio a causa de una dieta pobre ejerce sus efectos en los depósitos delhueso, más que en la concentración del calcio extracelular. En la actualidad, aún se ven insuficiencia de vitamina D en la dieta, a pesar de que muchos alimentos están enriquecidos con esta vitamina. Es muy probable que haya insuficiencia de vitamina D en estados de desnutrición, como obstrucción biliar, insuficiencia pancreática y enfermedad celíaca, en la que la capacidad de absorber grasa y vitaminas solubles en grasa está deteriorada.
La incapacidad de activar la vitamina D es otra causa de hipocalcemia. Los anticonvulsivos, sobre todo la fenitoína, afectan la activación inicial de la vitamina D en el hígado. El paso final de la activación de la vitamina D falla en personas con enfermedad renal crónica. Por suerte, la forma activada de vitamina D, calcitriol, se ha sintetizado y está disponible para utilizarse en el tratamiento de insuficiencia de calcio en personas con renopatía crónica.
Manifestaciones clínicas
La hipocalcemia se manifiesta como un trastorno agudo o crónico. Las manifestaciones de hipocalcemia aguda se reflejan en excitabilidad neuromuscular y efectos cardiovasculares incrementados por una disminución de calcio ionizado (tabla 39-10). El calcio ionizado estabiliza la excitabilidad neuromuscular, con lo que hace a las células nerviosas menos sensibles ante los estímulos. Los nervios expuestos a bajos niveles de calcio ionizado muestran umbrales menores para la excitación, respuestas repetidas a un solo estímulo y, en casos extremos, actividad continua.
La gravedad de las manifestaciones depende de la causa subyacente, rapidez de inicio, trastornos de los electrolitos acompañantes y pH extracelular. La excitabilidad neuromuscular incrementada se manifiesta como parestesias (es decir, hormigueo alrededor de la boca, y en manos y pies) y tetania (es decir, espasmos de los músculos del rostro, manos y pies). La hipocalcemia grave causa espasmos en la laringe, convulsiones e incluso la muerte.
Entre los efectos cardiovasculares de hipocalcemia aguda están hipotensión, insuficiencia cardíaca, arritmias cardíacas (sobre todo fibrilación ventricular y de las cavidades cardíacas) e insensibilidad a fármacos como digital, noradrenalina y dopamina, que actúan a través de mecanismos mediados por calcio.
Las pruebas de Chvostek y Trousseau se usan para valorar el incremento en la excitabilidad neuromuscular y tetania. El signo de Chvostek se provoca dando golpecitos en el rostro, justo abajo de la sien, en el punto donde sale el nervio facial. Los golpecitos en el rostro sobre el nervio facial causan espasmo del labio, nariz o rostro cuando el resultado de la prueba es positivo. En la prueba del signo de Trousseau se utiliza un manguito inflado para presión arterial; que se infla a 10 mm Hg por arriba de la presión sistólica durante 3 minutos. La contracción de dedos y manos (es decir, espasmo carpopedal) indica la presencia de tetania.
Con frecuencia, la hipocalcemia crónica está acompañada por manifestaciones en el esqueleto y cambios en la piel. Podría haber dolor de huesos, fragilidad, deformidades y fracturas. La piel puede estar seca y escamosa, la uñas quebradizas y el cabello seco. Es común la formación de cataratas.
Tratamiento
La hipocalcemia aguda es un caso de urgencia que requiere tratamiento expedito. Se administra una infusión intravenosa que contiene calcio (ej. gluconato de calcio y cloruro de calcio) cuando están presentes tetania o síntomas agudos o se prevén debido a reducción del nivel de calcio en plasma.
La hipocalcemia crónica se trata con ingesta de calcio. Un vaso de leche contiene alrededor de 300 mg de calcio. Se podrían administrar por vía oral complementos de calcio, de sales de carbonato, gluconato o lactato. El tratamiento de largo plazo podría necesitar preparaciones de vitamina D, sobre todo en personas con hipoparatiroidismo y renopatía crónica. La forma activa de vitamina D se administra cuando están dañados los mecanismos del hígado o los riñones necesarios para activar la hormona. La HPT (1-34) sintética se administra mediante inyecciones subcutáneas, como tratamiento de reemplazo en casos de hipoparatiroidismo.
Hipercalcemia
Este trastorno representa una concentración total de calcio plasmático mayor de 10,5 mg/dl (2,6 mmol/l). Niveles falsamente altos de calcio son resultado de extracciones prolongadas de sangre con un torniquete en exceso apretado. El aumento de proteínas en plasma (es decir, hiperalbuminemia y hiperglobulinemia) a veces incrementa el calcio total en plasma, pero no afecta las concentración de calcio ionizado.
Causas. Hay exceso de calcio plasmático (es decir, hipercalcemia) cuando el movimiento del calcio en la circulación satura las hormonas reguladoras de calcio o supera la capacidad del riñón para eliminar el exceso de iones de calcio (tabla 39-11). Las 2 causas más comunes de hipercalcemia son la reabsorción incrementada de hueso debida a neoplasias, e hiperparatiroidismo. Ambas causas se encuentran en la mayoría de las personas con hipercalcemia. Este trastorno es una complicación común de tumores, y se presenta en alrededor del 10% al 20% de personas con enfermedad avanzada; se denomina hipercalcemia tumoral (HCT). Una cantidad de tumores malignos, como el carcinoma pulmonar, se han vinculado con hipercalcemia. Algunos tumores destruyen el hueso, en tanto que otros generan agentes humorales que estimulan la actividad osteoclástica, aumentan la reabsorción del hueso, o bien, inhiben la formación de hueso.
La mayoría de pacientes con HCT producen una proteína relacionada con HPT (PrHPT), que es designada como el principal factor humoral que se encarga de la HCT. La HPT y PrHPT tiene notable semejanza, o similitud estructural, en sus extremos terminales amino. Esta similitud da como resultado que tanto HPT como HPT-rP se unen al mismo receptor (receptor HPT/PrHPT). El PrHPT se detecta en personas con muchos tipos de órganos sólidos y, también, con leucemia/linfoma de células T del adulto.
Las causas menos frecuentes de hipercalcemia son inmovilización prolongada, aumento de la absorción intestinal de calcio, dosis excesivas de vitamina D, o bien, los efectos de fármacos como litio y diuréticos con tiacida. Los niños con hipercalcemia necesitan excretar el calcio por la orina de manera expedita; esto es el principal objetivo del tratamiento. La inmovilización prolongada y la incapacidad para soportar peso generan desmineralización de los huesos y libera calcio en el torrente sanguíneo.
La absorción intestinal de calcio aumenta por las dosis excesivas de vitamina D o como resultado de una enfermedad llamada síndrome de leche y alcalinos. Este síndrome es causado por la ingesta excesiva de calcio (a menudo en forma de leche) y anti-ácidos absorbibles. Debido a la disponibilidad de antiácidos inabsorbibles, este trastorno ya se observa menos que en el pasado, pero podría presentarse en mujeres que son en exceso proclives a tomar preparaciones de calcio para prevenir la osteoporosis. La suspensión de los antiácidos repara la alcalosis y aumenta la eliminación de calcio.
Una diversidad de fármacos aumentan los niveles de calcio. El consumo de litio para tratar trastornos bipolares ha causado hipercalcemia e hiperparatiroidismo. Los diuréticos con tiacida incrementan la reabsorción de calcio en el túbulo contorneado distal de los riñones. Si bien los diuréticos con tiacida rara vez causan hipercalcemia, son capaces de ocultar ésta por otras causas, como trastornos óseos subyacentes y enfermedades que aumentan la reabsorción del hueso.
Manifestaciones clínicas
Los signos y síntomas relacionados con el exceso de calcio reflejan:
- cambios en al excitabilidad neural,
- alteraciones en la función del músculo liso y cardíaco, y
- exposición de los riñones a altas concentraciones de calcio (tabla 39-11).
La excitabilidad neural disminuye en pacientes con hipercalcemia. Podría haber embotamiento de la conciencia, estupor, debilidad y flacidez muscular. Los cambios en la conducta pueden ir desde alteraciones leves en la personalidad hasta psicosis agudas. El corazón responde a niveles altos de calcio con aumento de contractibilidad y arritmias ventriculares. El digital acentúa estas respuestas. Los síntomas gastrointestinales reflejan una disminución en la actividad del músculo liso, y entre otros, están estreñimiento, anorexia, náuseas y vómito. El alto contenido de calcio en la orina afecta la capacidad del riñón para concentrar orina, porque interfiere con la acción de HAD (un ejemplo de DI nefrógena). Esto causa diuresis de sal y agua, y un aumento en la sensación de sed. La hipercalciuria también predispone a la formación de cálculos renales. La pancreatitis es otra complicación potencial de la hipercalcemia y es probable que esté relacionada con cálculos en los conductos pancreáticos.
La crisis hipercalcémica describe un aumento agudo en el nivel del calcio plasmático. Los tumores y el hiperparatiroidismo son las causas principales de esta crisis. En ésta, las arritmias cardíacas, oliguria, sed excesiva, insuficiencia de volumen, fiebre, niveles alterados de conciencia y un estado mental trastornado se presentan junto con otros signos característicos de exceso de calcio. La hipercalcemia sintomática se relaciona con alta mortalidad y, con frecuencia, la muerte es causada por paro cardíaco.
Tratamiento
Por lo general, el tratamiento para el exceso de calcio se enfoca en la rehidratación y aplicación de medidas para incrementar la excreción urinaria de calcio. Es necesario el reemplazo de líquido en situaciones de insuficiencia de volumen. La excreción de sodio va junto con la excreción de calcio. Los diuréticos y el NaCl se administran para aumentar la eliminación urinaria de calcio después de que se restaura el volumen de LEC. Por lo general, se utilizan los diuréticos de asa y no los de tiacida, que aumentan la reabsorción de calcio. La reducción inicial en los niveles de calcio es seguida por medidas para inhibir la reabsorción de hueso. Entre los fármacos que se administran para inhibir la movilización de calcio están bisfosfonatos, calcitonina, corticoesteroides, mitramicina y nitrato de galio. Los bisfosfonatos (ej. pamidronato y zoledronato), cuya acciónprincipal consiste en inhibir la actividad osteoclástica, consiguen una reducción considerable de los niveles de calcio con relativamente pocos efectos secundarios. La calcitonina inhibe la actividad osteoclástica, con lo que disminuye la reabsorción. Los corticoesteroides y mitramicina inhiben la reabsorción de hueso y se administran para tratar hipercalcemia asociada con cáncer.
El consumo prolongado de mitramicina, un antineoplásico, está limitado, debido a su potencial de nefrotoxicidad y hepatotoxicidad. El nitrato de galio es muy efectivo para tratar hipercalcemia grave secundaria tumor. Es un compuesto químico que inhibe la reabsorción ósea, aunque se desconoce el mecanismo preciso de acción. También se puede aplicar diálisis en paciente con hipercalcemia e insuficiencia renal, y en personas con insuficiencia cardíaca, en quienes la sobrecarga de líquidos es un problema.
Trastornos del equilibrio del fósforo
El fósforo es sobre todo un anión intracelular. Alrededor del 85% de fósforo está contenido en los huesos y la mayor parte del resto (14%) se encuentra en las células. Sólo casi el 1% está en el compartimento del LEC y de éste sólo una proporción mínima está en el plasma. En el adulto, el nivel de fósforo está entre 2,5 mg/dl y 4,5 mg/dl (0,8 mmol/l a 1,45 mmol/l). Estos valores son ligeramente superiores en los lactantes (3,7 mg/dl a 8,5 mg/dl, 01,2 mmol/l a 02,7 mmol/l) y niños (4 mg/dl a 5,4 mg/dl, 1,3 mmol/l a 1,7 mmol/l), tal vez debido a que hay más hormona del crecimiento y menos hormonas de las gónadas.
El fósforo existe en 2 formas dentro del cuerpo: inorgánico y orgánico. La inorgánica (fosfato [H2PO4-, o bien, HPO42-]) es la principal forma circulante de fósforo y es la que se mide de manera obligada (y se anota como fósforo) en los estudios de laboratorio. La mayor parte de fósforo intracelular (alrededor del 90%) está en forma orgánica (ej. ácidos nucleicos, fosfolípidos, ATP).
La entrada de fósforo a las células mejora después de consumir glucosa, porque el fósforo está incorporado en los intermediarios fosforilados del metabolismo de la glucosa. Las lesiones en las células o la atrofia celular causan una pérdida de componentes celulares que contienen fosfato orgánico; la regeneración de estos componentes celulares origina el retiro de fosfato inorgánico del compartimento del LEC.
El fósforo es esencial en muchas de las funciones corporales. Tiene un papel principal en la formación del hueso; es esencial en ciertos procesos metabólicos como la formación de ATP y las enzimas necesarias para el metabolismo de la glucosa, grasas y proteínas; es un componente indispensable de varias partes vitales de la célula, ya que está incorporado en los ácidos nucleicos de ADN y ARN, y en los fosfolípidos de la membrana celular, y funciona como un amortiguador acidobásico en el LEC y en la excreción renal de iones hidrógeno. El abastecimiento de O2 que realizan los glóbulos rojos depende del fósforo orgánico en el ATP y 2,3-difosfoglicerato (2,3-DFG). Además, el fósforo es necesario para el funcionamiento normal de otros elementos formes de la sangre, como los glóbulos blancos y las plaquetas.
Ganancias y pérdidas
El fósforo se ingiere con la dieta y se elimina con la orina. Proviene de muchas fuentes de la dieta, como leche y carnes. Casi el 80% del fósforo ingerido es absorbido en el intestino, sobre todo en el yeyuno. La absorción disminuye cuando se ingieren a la vez sustancias que se unen al fósforo, como calcio, magnesio y aluminio.
El fosfato no se une a las proteínas plasmáticas y, en esencia, todo el que está presente en el plasma es filtrado en el glomérulo. La eliminación del fosfato en los riñones está regulada entonces por un mecanismo de rebosamiento en el que la cantidad de fosfato perdido con la orina estárelacionado de manera directa con las concentraciones de fosfato en la sangre. De hecho, todo el fosfato filtrado es reabsorbido cuando son bajos los niveles de éste; cuando las concentraciones de fosfato plasmático suben por arriba del nivel crítico, el exceso de fosfato es eliminado con la orina.
El fosfato es reabsorbido desde el filtrado en las células epiteliales del túbulo proximal, a través de la acción de un cotransportador (NPT2, por sus siglas en inglés) de sodiofosfato. La HPT desempeña una función significativa al regular la reabsorción del fosfato inhibiendo la síntesis y la expresión del transportador de NPT2. Por consiguiente, siempre que HPT aumenta, disminuye la reabsorción tubular de fosfato y éste se pierde más con la orina.
El NPT2 también es inhibido por las hormonas llamadas fosfatoninas. Las fosfatoninas más importantes son 2:
- factor de crecimiento de fibroblastos 23 (FCF 23), y
- la proteína secretada 4 relacionada con frizzled (sFRP4, por sus siglas en inglés).
Cuando hay una producción excesiva de estas hormonas, como en el caso de osteomalacia inducida por tumor, hay hipofosfatemia muy marcada, debido a una menor absorción de fosfato intestinal. Además, el aumento de fosfatonina causa una degradación excesiva de calcitriol (vitamina D activa), lo que deriva en osteomalacia.
Hipofosfatemia
En general se define por una concentración de fósforo plasmático menor a 2,5 mg/dl (0,8 mmol/l) en adultos; se considera grave cuando la concentración en menor de 1 mg/dl (0,32 mmol/l). Puede haber hipofosfatemia a pesar de los depósitos de fosfato normales en el cuerpo, como resultado del movimiento desde el compartimento del LEC al LIC. Puede haber insuficiencia grave de fósforo con concentraciones plasmáticas bajas, normales o altas.
Causas
Las causas más comunes de hipofosfatemia son insuficiencia de fósforo causada por absorción intestinal deteriorada, cambios compartimentales y aumento de las pérdida renales (tabla 39-12). Con frecuencia, más de uno de estos mecanismos está activo. A menos que la ingesta de alimentos sea restringida de manera rigurosa, la ingesta en la dieta y la absorción intestinal de fósforo son adecuadas. La absorción intestinal se puede inhibir mediante administración de glucocorticoides, niveles de magnesio muy altos en la dieta e hipotiroidismo. La ingesta prolongada de antiácidos también podría interferir con la absorción intestinal. Los antiácidos que contienen hidróxido de aluminio, carbonato de aluminio y carbonato de calcio se unen al fosfato, lo que causa un aumento en las pérdidas de fosfato con las heces. Debido a su capacidad para unirse al fosfato, a veces los antiácidos elaborados con calcio se usan terapéuticamente para reducir las concentraciones de fosfato en plasma en las personas con renopatía crónica.
El alcoholismo es una causa común de hipofosfatemia. El mecanismo subyacente de la hipofosfatemia en la persona adicta al alcohol podría estar relacionado con desnutrición, aumento en los índices de excreción renal o hipomagnesemia. La desnutrición y la cetoacidosis diabética incrementan la excreción de fosfato y la pérdida de fósforo del cuerpo. Al alimentar a los pacientes desnutridos aumenta la incorporación de fósforo a los ácidos nucleicos y a los compuestos fosforilados de la célula. Lo mismo sucede cuando la cetoacidosis diabética se revierte con el tratamiento de insulina. Las pérdidas de fosfato en la orina pueden ser causadas por fármacos, como teofilina, corticoesteroides y diuréticos de asa, que incrementan la excreción renal.
La hipofosfatemia también se presenta durante la administración prolongada de glucosa o hiperalimentación, mientras que la de laglucosa causa liberación de insulina, con transporte de glucosa y fósforo hacia la célula. Los fenómenos catabólicos que se dan en la cetoacidosis diabética también agotan los depósitos de fósforo. Por lo general, la hipofosfatemia no es evidente sino hasta que la insulina y el reemplazo de líquidos revierten la deshidratación y la glucosa empieza a retroceder hacia dentro de las células. La administración de soluciones para hiperalimentación sinfósforo suficiente ocasiona un rápido flujo de fósforo hacia dentro de la masa muscular del cuerpo, en particular si el tratamiento inicia después de un período de catabolismo del tejido. Puesto que sólo una pequeña cantidad del fósforo corporal total está en el compartimento del LEC, incluso una pequeña redistribución entre los compartimentos del LEC y del LIC es capaz de causar hipofosfatemia, aun cuando no hayan cambiado los niveles totales de fósforo.
La alcalosis respiratoria debida a hiperventilación prolongada origina hipofosfatemia por niveles reducidos de calcio ionizado a causa del aumento de la unión con proteínas, mayor liberación de HPT y mayor excreción de fosfato.
Manifestaciones clínicas
Las manifestaciones de insuficiencia de fósforo se producen porque hay un decremento de depósitos de energía celulares debido a la insuficiencia de ATP y a transporte insuficiente de O2, a causa de menos 2,3-DPFG de los glóbulos rojos. La hipofosfatemia genera alteraciones en la función neural y musculoesquelética, además de trastornos hematológios (tabla 39- 12).
El metabolismo de los glóbulos rojos se daña por la insuficiencia de fósforo; las células se vuelven rígidas, sufren mayor hemólisis y tienen menos ATP y 2,3-DPFG. También se afectan las funciones quimiotácticas y fagocíticas de los glóbulos blancos y las funciones hemostáticas de las plaquetas. La hipofosfatemia grave aguda (0,1 mg/dl a 0,2 mg/dl) causa anemia hemolítica aguda, con un aumento en la fragilidad de los eritrocitos, mayor susceptibilidad a la infecciones y disfunción de plaquetas, con hemorragias petequiales. Puede haber anorexia y disfagia. Las manifestaciones neurales (temblor intencional, parestesias, hiporreflexia, estupor, coma y convulsiones), aunque raras, son manifestaciones de gravedad. La insuficiencia respiratoria que resulta de función deteriorada de los músculos respiratorios se presenta en personas con hipofosfatemia grave.
La insuficiencia crónica de fósforo interfiere con la mineralización de matriz ósea recién formada. En los niños en crecimiento, este proceso provoca crecimiento endocondral anómalo y manifestaciones clínicas de raquitismo. En los adultos, el trastorno ocasiona rigidez en las articulaciones, dolor de huesos y deformidades esqueléticas consistentes con osteomalacia.
Tratamiento
Por lo general, el tratamiento se centra en la profilaxis. Se puede lograr con dieta alta en fósforo (un vaso de leche contiene alrededor de 250 mg de fósforo) o con soluciones de reemplazo por vía oral o intravenosa. En casos de hiperparatiroidismo, renopatía crónica e hipercalcemia es común que los complementos de fósforo estén contraindicados, dado el mayor riesgo de calcificaciones extracelulares.
Hiperfosfatemia
Representa una concentración plasmática de fósforo por arriba de 4,5 mg/dl (1,45 mmol/l) en adultos. Los niños en crecimiento tienen, por lo regular, niveles de fosfato plasmático superiores a los de los adultos.
Causas
La hiperfosfatemia es resultado de la incapacidad de los riñones para excretar el exceso de fosfato y de la redistribución rápida de fosfato intracelular al compartimento del LEC, además del consumo excesivo de fósforo. La causa más común es una deficiencia de la función renal (tabla 39-13).
La hiperfosfatemia es un trastorno común de los electrolitos en personas con renopatía crónica, en quienes se presenta un aumento en los niveles de fósforo, a pesar de los aumentos compensadores de HPT. La evidencia señala un incremento de calcificación cardiovascular y mortalidad en personas con renopatía crónica y concentraciones elevadas de fósforo. La liberación de fósforo intracelular deriva de trastornos como lesión masiva de tejidos, rabdomiólisis, accidente cerebrovascular, insuficiencia de potasio y convulsiones. La quimioterapia es capaz de aumentar las concentraciones de fosfato plasmático, dada la rápida destrucción de células tumorales (síndrome de lisis tumoral).
La administración excesiva de antiácidos que contienen fosfato, laxantes o enemas es otra causa de hiperfosfatemia, en especial cuando existe un decremento en el volumen vascular y un índice bajo de filtración glomerular. Por la inducción de diarrea, los laxantes y enemas que contienen fosfato predisponen a hipovolemia y a un índice bajo de filtración glomerular, lo que aumenta el riesgo de hipofosfatemia.
Manifestaciones clínicas
La hiperfosfatemia va paralela a un decremento del calcio plasmático.
Muchos de los signos y síntomas de un exceso de fosfato están relacionados con una insuficiencia de calcio (tabla 39-13). El tratamiento incorrecto de la hiperfosfatemia en enfermedad crónica causa hiperparatiroidismo secundario, osteodistrofias renales o trastornos minerales en hueso y calcificaciones extraóseas en tejidos lisos.
Tratamiento
El tratamiento de hiperfosfatemia se centra en la causa del trastorno. Se pueden aplicar restricciones de alimentos con alto contenido de fósforo. Los enlazantes de fosfato elaborados con calcio son útiles en la hiperfosfatemia crónica. El sevelamer, un enlazante de fosfato sin calcio ni aluminio, es un enlazante tan efectivo como uno elaborado con calcio y no provoca manifestaciones adversas secundarias como aumento del producto calcio × fosfato, hipercalcemia y calcificaciones vasculares y cardíacas. En personas con renopatía crónica se aplica hemodiálisis para disminuir la concentración de fosfato.
Trastornos del equilibrio del magnesio
El magnesio ocupa el cuarto lugar entre los cationes abundantes en el cuerpo y el segundo entre los cationes intracelulares; el primero es del potasio. Del total del contenido de magnesio, alrededor del 50% al 60% está depositado en hueso, del 39% al 49% está en las células corporales y el restante 1% está disperso en el LEC. Entre el 20% y el 30% del magnesio del LEC está unido a proteínas, y sólo una pequeña fracción del magnesio del LIC (del 15% al 30%) es intercambiable con el LEC. En plasma, la concentración normal de magnesio es 1,8 mg/dl a 3,0 mg/dl (0,75 mmol/l a 1,25 mmol/l).
Hace apenas poco tiempo que se reconoció la importancia de magnesio en todas las funciones del cuerpo. El magnesio actúa como cofactor en muchas de las reacciones enzimáticas intracelulares, como la transferencia de grupos fosfato de alta energía en la generación de ATP a partir difosfato de adenosina (ADP). Es esencial en todas las reacciones que requieren ATP, en cada etapa relacionada con la copia y transcripción del ADN y para la traducción del ARN mensajero. Se requiere en el metabolismo de la energía en la célula, funcionamiento de la bomba de membrana Na+/K+-ATPasa, estabilización de la membrana, conducción en nervios, transporte de iones y actividad de los canales del potasio y el calcio.
Los canales del potasio, incluso el canal del potasio sensible a la acetilcolina, dependen de niveles intracelulares correctos. El magnesio bloquea el movimiento de salida del potasio en las células cardíacas. Cuando el nivel de magnesio es bajo, el canal permite el flujo la salida del potasio, lo que causa bajos niveles de éste dentro de la célula. Muchos canales del calcio también dependen del magnesio. Concentraciones altas de magnesio en el LIC inhiben el transporte del calcio hacia las células y su liberación desde el retículo sarcoplásmico. Por tanto, el magnesio tiende a actuar como un relajante del músculo liso, al modificar los niveles de calcio que son los encargados de la contracción muscular.
El magnesio tiene un efecto anticonvulsivo. El mecanismo de acción sugerido es vasodilatación cerebral o prevención de daño neuronal isquémico mediante bloqueo de los receptores de N-metil-D-aspartato (NMDA) en el cerebro. El magnesio es la medicación de primera línea para tratar eclampsia en las mujeres embarazadas. Además, a menudo se emplea como neuroprotector en los lactantes. En efecto, la evidencia indica que, cada año, se podrían evitar cerca de 1.000 casos de parálisis cerebral en Estados Unidos si durante el trabajo de parto se usara magnesio en forma consistente.
Ganancias y pérdidas
El magnesio se ingiere con la dieta, es absorbido en el intestino y excretado por los riñones. La absorción intestinal no está regulada estrechamente, por lo que se absorbe alrededor del 25% al 65% del magnesio de la dieta. El magnesio está contenido en todos los vegetales verdes, cereales, nueces, pescados y mariscos. También está presente en gran parte del agua subterránea de Estados Unido y Canadá.
El riñón es el principal órgano de regulación del magnesio. Filtra alrededor de 70% a 80% del magnesio plasmático y excreta casi el 6%, si bien esta cantidad puede estar determinada por otros trastornos y medicaciones. El magnesio es el único electrolito del que sólo cerca del 12% al 20% de la cantidad filtrada es reabsorbida en el túbulo proximal. La mayor cantidad, casi el 70%, es reabsorbida de manera pasiva en la rama ascendente gruesa del asa de Henle. La principal fuerza activadora para la absorción del magnesio en la rama ascendente gruesa del asa de Henle es el gradiente de voltaje positivo creado en el lumen del túbulo por el sistema de cotransporte de Na+/K+/2Cl+. La inhibición de este sistema de transporte que realizan los diuréticos de asa reduce la reabsorción de magnesio. La reabsorción activa de éste tiene lugar en el túbulo contorneado distal y representa casi el 10% de la carga filtrada. La HPT estimula la reabsorción de magnesio, y ésta disminuye en presencia de niveles plasmáticos altos de magnesio y calcio.
Hipomagnesemia
La insuficiencia de magnesio se refiere al agotamiento de los depósitos corporales totales, en tanto que la hipomagnesemia es una concentración de magnesio plasmático inferior a 1,8 mg/dl (0,75 mmol/l). Se detecta en situaciones en que se limita la ingesta o hay aumento de pérdidas intestinales o renales; es un hallazgo común en las salas de urgencia y en unidades de cuidado intensivo.
Causas
La insuficiencia de magnesio es resultado de ingesta insuficiente, pérdidas excesivas, o bien, movimiento entre los compartimentos del LEC y del LIC (tabla 39-14). Deriva de trastornos que restringen la ingesta de manera directa, como desnutrición, inanición o mantenimiento prolongado de nutrición parenteral sin magnesio y de otros trastornos, como diarrea, síndromes de malabsorción, succión nasogástrica prolongada, exceso de laxantes o menor absorción intestinal.
Otra causa común de insuficiencia de magnesio es el alcoholismo crónico. Muchos factores contribuyen a hipomagnesemia en el alcoholismo, incluso baja ingesta y pérdidas gastrointestinales por diarrea. Los efectos de hipomagnesemia aumentan de modo exagerado con otros trastornos electrolíticos, como hipocaliemia, hipocalcemia y acidosis metabólica.
Si bien los riñones son capaces de defenderse contra la hipermagnesemia, son menos capaces de conservar magnesio y evitar hipomagnesemia. Las pérdidas por la orina aumentan en la cetoacidosis diabética, hiperparatiroidismo e hiperaldosteronismo. Algunas sustancias incrementan las pérdidas renales de magnesio, como los diuréticos de asa y con tiacida, y fármacos nefrotóxicos como antibióticos aminoiglucósidos, ciclosporina, cisplatina y anfotericina B. Varias enfermedatdes genéticas raras producen hipomagnesemia (ej. síndromes de Gitelman y Bartter).
También podría haber hipomagnesemia relativa en situaciones que propicien el movimiento de magnesio entre los compartimentos del LEC y del LIC, como administración rápida de glucosa, soluciones parenterales que contienen insulina y alcalosis. Aunque son momentáneas, estas situaciones ocasionan graves alteraciones en las funciones corporales.
Manifestaciones clínicas
La insuficiencia de magnesio se presenta junto con hipocalcemia e hipocaliemia, lo que provoca una cantidad de manifestaciones neurológicas y cardiovasculares relacionadas (tabla 39-14). La hipocalcemia es característica de hipomagnesemia grave. La mayoría de personas con hipocalcemia relacionada con hipomagnesemia tiene niveles reducidos de HPT, quizá como resultado de mecanismos dañados que dependen de magnesio y que controlan la liberación y síntesis de HPT. También hay evidencia de que la hipomagnesemia disminuye tanto la liberación de calcio del hueso dependiente de HPT como la independiente de HPT. En la hipomagnesemia, los iones de magnesio (Mg2+) se liberan desde el hueso en intercambio por la captación incrementada de calcio desde el LEC.
La hipomagnesemia causa una reducción de potasio intracelular y deteriora la capacidad de los riñones para conservarlo. Cuando hay hipomagnesemia, la hipocaliemia es insensible al tratamiento de reemplazo de potasio.
El magnesio es vital para el metabolismo de los carbohidratos y la generación tanto del metabolismo aeróbico como anaeróbico. Muchas de las manifestaciones de déficit de magnesio son causadas por trastornos electrolíticos relacionados, como hipocaliemia e hipocalcemia. Esta última se podría probar mediante la existencia de cambios en la personalidad e irritabilidad neuromuscular junto con temblor, movimientos atetoides o coreiformes y signos de Chvostek y Trousseau positivos.
Entre las manifestaciones cardiovasculares están taquicardia, hipertensión y arritmias ventriculares. Podría haber cambios en la ECG, como ampliación del complejo QRS, aparición de ondas T máximas, prolongación del intervalo PR, inversión de la onda T a surgimiento de ondas U. Las arritmias ventriculares, sobre todo en presencia de digital, podrían ser difíciles de tratar, a menos quese normalicen los niveles de magnesio.
Se ha considerado que la insuficiencia persistente de magnesio es un factor de riesgo para osteoporosis y osteomalacia, en particular en personas con alcoholismo crónico, diabetes mellitus y síndrome de malabsorción.
Tratamiento
Se trata con reemplazo de magnesio. La vía de administración depende de la gravedad del trastorno. La insuficiencia de magnesio sintomática, moderada a grave, se trata con la administración parenteral. El tratamiento debe continuar durante varios días para reemplazar las concentraciones almacenadas y plasmáticas. Cuando la pérdida intestinal o renal es crónica, podría ser necesario el tratamiento de apoyo con magnesio por vía oral. Con frecuencia, el magnesio se administra terapéuticamente para tratar arritmia cardíaca, infarto de miocardio, angina, asma bronquial y embarazo complicado por preeclampsia o eclampsia. Es esencial la precaución para evitar hipermagnesemia; es importante vigilar con todo cuidado a las personas con cualquier grado de insuficiencia renal para evitar el exceso de magnesio.
Hipermagnesemia
Representa un aumento en el magnesio total del cuerpo y una concentración de magnesio plasmático por arriba de 3,0 mg/dl (1,25 mmol/l). La hipermagnesemia es una alteración rara, porque el riñón normal tiene la capacidad de excretar magnesio.
Causas
Cuando se presenta hipermagnesemia, por lo general, está relacionada con insuficiencia renal y el consumo imprudente de medicamentos que contienen magnesio, como antiácidos, complementos minerales o laxantes (tabla 39-15).
Los adultos mayores están particularmente en riesgo, porque sus funciones renales disminuyen al avanzar la edad y tienden a consumir más medicamentos que contienen magnesio, como antiácidos y laxantes. El sulfato de magnesio se utiliza para tratar la toxemia en el embarazo y trabajo de parto prematuro; en estos casos es esencial la vigilancia en busca de signos de hipermagnesemia.
Manifestaciones clínicas
La hipermagnesemia afecta la función neuromuscular y cardiovascular (tabla 39-15). Puesto que el magnesio tiende a inhibir la secreción de HPT, podría haber hipocalcemia e hipermagnesemia. Por lo general, los signos y síntomas se presentan sólo cuando los niveles de magnesio plasmático sobrepasan 4,8 mg/dl (2 mmol/l).
La hipermagnesemia disminuye la función neuromuscular, lo que ocasiona hiporreflexia, debilidad muscular y confusión. El magnesio reduce la liberación de acetilcolina en la articulación neuromuscular y podría generar bloqueo neuromuscular y parálisis respiratoria. Los efectos cardiovasculares se relacionan con los efectos bloqueadores de los canales del calcio que ejerce el magnesio. La presión arterial es baja y la ECG muestra un acortamiento del intervalo QT, anomalías en la onda T y prolongación de los intervalos QRS y PR. La hipermagnesemia grave (>12 mg/dl) se relaciona con parálisis muscular y respiratoria, bloqueo cardíaco completo y paro cardíaco.
Tratamiento
Entre otras prácticas, está la suspensión de la administración de magnesio. El calcio es un antagonista directo del magnesio, por lo que se podría administrar por vía intravenosa. Podría ser necesario aplicar diálisis o hemodiálisis peritoneal.