Patología fetal. Clasificación y etiología
La patología fetal comprende el conocimiento de los estados patológicos que cursan o se originan antes de que tenga lugar el nacimiento a la vida extrauterina, y que obedecen a dos grandes categorías de factores etiológicos: genéticos o endógenos y ambientales o exógenos. En ocasiones ambos actúan de forma aislada, pero frecuentemente lo hacen de manera conjunta, como pueden ser los casos de malformaciones congénitas originadas por herencia multifactorial. Numerosos cuadros permanecen sin estar adscritos a una etiología conocida. Las causas de las malformaciones congénitas pueden establecerse aproximadamente en la mitad de los casos. Nelson y Holmes encontraban una frecuencia del 2,2% de malformaciones en el RN; el 10% de origen genético (anomalías cromosómicas, genes mutantes únicos, casos familiares), el 23% por herencia multifactorial, el 3,2% producidas por agentes teratógenos, el 2,5% debidas a factores uterinos, el 0,4% asociadas a gemelaridad y el restante de etiología desconocida. Un estudio reciente realizado en los Estados Unidos identifica una causa genética subyacente (única o multifactorial) en el 71% de los ingresos infantiles, que llega a alcanzar el 96% en el caso de las enfermedades crónicas. El gasto sanitario de su asistencia representó el 81% del presupuesto asistencial total. En la actualidad se reconoce que aproximadamente el 10% de anomalías estructurales congénitas son el resultado del efecto adverso de factores ambientales que actúan durante el desarrollo prenatal.
Epidemiología
Un estudio del Registro de Malformaciones en España, sobre una serie de 137.760 nacimientos consecutivos, obtiene una incidencia de malformaciones congénitas del 2,66%, algo superior a la citada antes. Las malformaciones más frecuentes son las cardiovasculares (11,6‰), seguidas de las de miembros y osteoarticulares (6,68‰) y urogenitales (6,2‰).
La incidencia general de los defectos estructurales congénitos se estima entre el 0,5 y el 1% de la población; si se utilizan los diagnósticos de alta de un hospital esta frecuencia asciende al 2-4% y si se vigila durante varios años el desarrollo de los niños y se incluyen los defectos menores, la frecuencia total puede alcanzar hasta el 10%.
En el Departamento de Pediatría de la Universidad de Zaragoza se encontró una incidencia de malformaciones congénitas del 1,73%. El riesgo de recurrencia de malformaciones congénitas, la posibilidad de su diagnóstico prenatal e, incluso, su tratamiento, han originado en los últimos años notables controversias y dilemas éticos, que comenzaron en 1982 con el caso de un RN afecto de un síndrome de Down con malformaciones digestivas asociadas que falleció sin haber sido intervenido. Por otro lado, constituyen la principal causa de mortalidad infantil, alcanzando tasas del 20-30% de todos los fallecimientos.
Un importante número de supervivientes pueden sufrir una inferioridad funcional (daño neurológico o trastornos conductuales, principalmente) o tener un lugar de menor resistencia sobre el que inciden patologías sobreañadidas.
Clasificación
Puede sistematizarse la patología prenatal de acuerdo con el momento de la gestación en que actúa el agente patógeno. Tradicionalmente se agrupan en cuatro grandes grupos: gametopatías, blastopatías, embriopatías y fetopatías.
Gametopatías. La noxa patógena actúa sobre los gametos, por lo que, generalmente, se trata de patología preconcepcional. Este grupo se subdivide en:
- cromosomopatías: errores cromosómicos numéricos y estructurales;
- genopatías: enfermedades hereditarias, resultantes de mutaciones puntuales de largo alcance presentes en el oocito o espermatocito, o en ambos a la vez, siendo transmitidas a la descendencia de acuerdo con las leyes de herencia mendeliana.
Blastopatías. Engloban cuadros patológicos surgidos en el periodo de blástula o primeros 18-21 días del desarrollo. Desde la fertilización hasta el periodo postimplantación temprano, el nuevo ser posee relativamente pocas células y está dotado de una gran capacidad para reponer las células pluripotenciales. Por ello, el efecto de un agente tóxico sigue el fenómeno del “todo o nada”: es decir, o se produce la lesión de un gran número de células (muerte del producto), o se afectan tan pocas que, generalmente, tiene lugar la reparación sin consecuencias lesionales. Durante la tercera semana de gestación, sin embargo, los efectos tóxicos sobre el producto pueden originar monstruosidades.
Embriopatías. Corresponden a la patología del embrión y están caracterizadas clínicamente por desviaciones del desarrollo de los órganos, dando lugar a malformaciones congénitas, únicas o múltiples. Cronológicamente quedan delimitadas al periodo que se inicia durante la cuarta semana del desarrollo y que finaliza en la 12ª semana de la gestación.
Fetopatías. Son las enfermedades del periodo fetal, que comienza al final de la 12ª semana y termina con el nacimiento. Algunos clínicos añadieron una última categoría denominada kiemopatías, para abarcar los trastornos prenatales debidos a patología placentaria.
El genotipo obedece a la carga genética suministrada al cigoto por los gametos masculino y femenino. El fenotipo es la resultante de este informe genético modificado por diversos agentes ambientales (epigenética). Las gametopatías tienen un origen generalmente endógeno, aunque pueden ser influenciadas por circunstancias ambientales. Las blastopatías constituyen un grupo etiológicamente mal conocido; en algunos casos (mosaicismos cromosómicos) obedecen a factores endógenos, pero verosímilmente los factores causales serán ambientales. Embriopatías y fetopatías son de origen exógeno o ambiental, si bien en distintos casos estos factores permanecen sin aclarar.
Estos factores causales pueden actuar aisladamente pero, en otras ocasiones, precisan para manifestarse de condiciones genéticas predisponentes. Esta sistemática clásica tiene utilidad clínica, si bien su delimitación cronológica es objeto de discusión. En muchas ocasiones no resulta fácil establecer una neta diferencia entre embriopatías o fetopatías; asimismo, el agente etiológico puede persistir en su actuación durante ambos periodos (virus de la rubéola). Por esta razón, la OMS utiliza el término de embriofetopatía para designar la totalidad de la patología prenatal, definiéndola como toda alteración congénita, morfológica, biológica o conductual, inducida en cualquier momento de la gestación y detectada en el momento del nacimiento o más tardíamente. También se acepta la más amplia denominación de enfermedades fetales.
Características de la etapa prenatal
Cada periodo prenatal posee características específicas, que van a condicionar la expresión de los distintos cuadros clínicos.
Periodo de blástula
Es definido morfológicamente por la formación de las tres hojas blastodérmicas; la nutrición se realiza por difusión y la reacción a la agresión se traduce en forma de desplazamiento de masas o regulación. La expresión clínica, como se dijo, es la muerte del producto o bien la aparición de grandes monstruosidades.
El fenómeno más característico que tiene lugar durante la 3a semana es la gastrulación. La persistencia de restos de la línea primitiva en la región sacrocoxígea, puede originar tumores que contienen tejidos derivados de las tres capas germinativas (teratomas sacrocoxígeos).
Periodo embrionario
En él ocurre la formación de los distintos órganos y tejidos (organogénesis). La nutrición se realiza a partir de la circulación sanguínea. La reacción del producto frente a la agresión tiene lugar en forma de fenómenos de degeneración y regeneración locales, que patológicamente se traducen en forma de malformaciones congénitas de las estructuras que están en un periodo crítico de su desarrollo.
Periodo fetal
Se caracteriza por el crecimiento y desarrollo de las estructuras previamente formadas. La nutrición continúa siendo sanguínea. El feto reacciona a la agresión en forma de respuestas ordenadas similares a las que tienen lugar durante la vida postnatal. Este periodo se denomina también de histogénesis. Los agentes teratógenos pueden disminuir la población celular al originar muerte celular, retraso de su crecimiento o inhibición de la diferenciación celular. Clínicamente los procesos patológicos se expresan en forma de lesiones tisulares. La reacción del huésped ante la agresión depende del momento del desarrollo en que tenga lugar. De acuerdo con el denominado “periodo crítico” de cada órgano y tejido, su vulnerabilidad será variable. El momento vulnerable crítico para un determinado sistema coincide con la máxima intensidad de síntesis de ADN. Durante las dos primeras semanas del periodo de blástula, ésta no es vulnerable por los agentes patógenos (factores teratógenos) en orden a producir malformaciones. Sin embargo, una vez superado dicho momento cronológico, se inicia una etapa crítica de producción de malformaciones.
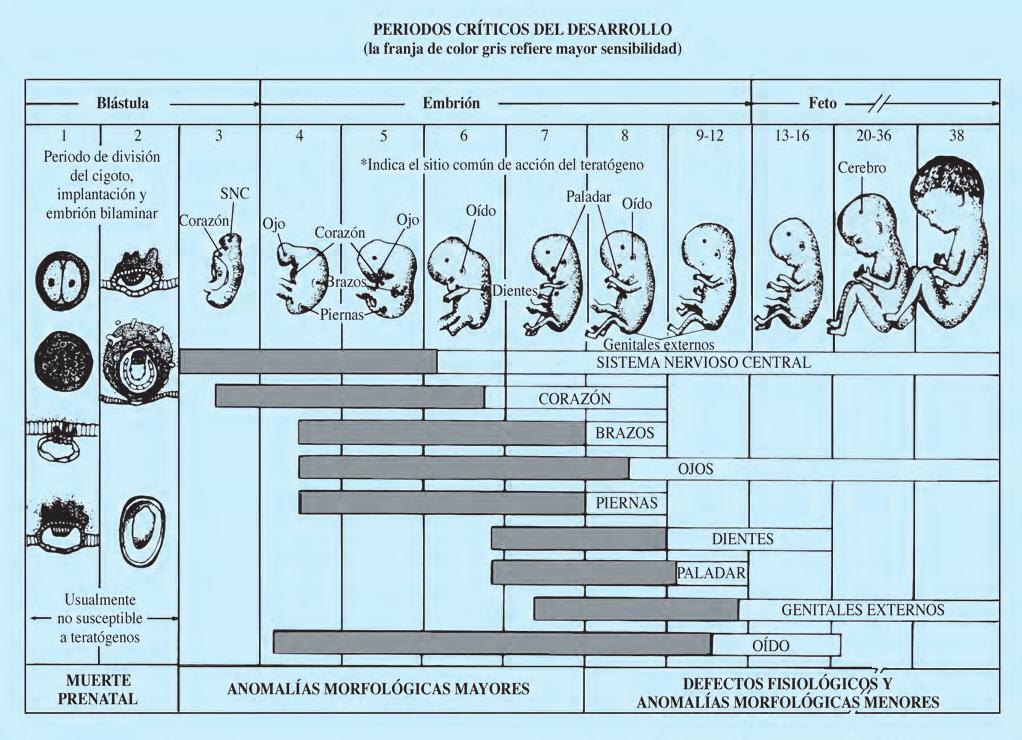
De acuerdo con el esquema de Moore de los estadios críticos del desarrollo (Fig. 2.6.2), se establece la época predominante de inducción de anomalías morfológicas mayores y menores. El lugar del organismo embrionario donde selectivamente actúa el agente teratógeno queda bien establecido. Durante el desarrollo fetal las anomalías morfológicas son menores y hacen su aparición los defectos funcionales o fisiológicos. El referido esquema evidencia el factor cronoespecificidad, que hace más importante el momento en el que se produce la agresión al producto que la propia naturaleza del factor causal. Derivado del anterior concepto ha podido establecerse un auténtico “calendario de malformaciones”, como ejemplariza la embriopatía rubeólica: hacia el final del primer mes de gestación aparecen principalmente catarata y cardiopatía; durante el segundo mes se mantiene inicialmente el riesgo de producción de cardiopatía y, a lo largo de todo él, el daño cerebral; la sordera es característica de la infección del embrión durante el tercer mes.
Etiología general
Actúan tanto factores genéticos como ambientales.
Factores genéticos
Son responsables de tres importantes grupos de enfermedades fetales: enfermedades monogénicas, desórdenes multifactoriales y cromosomopatías.
Recientemente se han incorporado otros nuevos, como la herencia mitocondrial, la disomía uniparental, el fenómeno de anticipación genética y las mutaciones celulares somáticas, lo que permite, por ejemplo, explicar la estrecha relación entre malformación y cáncer. Los defectos cromosómicos se asocian a malformaciones congénitas múltiples, tanto más graves cuanto mayor sea el imbalance genético (aneuploidías, triploidías). El accidente de la no disyunción suele ser el responsable de los síndromes de trisomía, que son las anomalías mejor conocidas, lo que se relaciona, en parte, con la edad materna avanzada. No obstante, el uso del concepto “fracción atribuible” formula interrogantes al factor edad materna. Aquel término se refiere a la fracción de casos de una enfermedad determinada en una población que podrían haberse evitado si el factor de riesgo no hubiese estado presente. Según Khoury, sólo un 20% de nacimientos de niños con síndrome de Down es atribuible a los grupos de edad materna superior a los 35 años. De ahí que persista el nacimiento de niños trisómicos cuando el screening sólo se hace en gestantes mayores.
La incorporación de las modernas técnicas de cariotipo de alta resolución ha permitido identificar un gran número de anomalías, dentro de las que destacan los denominados síndromes de microdeleción, mientras la patología molecular, con la incorporación de la tecnología ADN recombinante, permite en la actualidad explicar fenómenos tales como los clásicos conceptos de penetrancia y expresividad, o argumentar el porqué aparentemente una misma microdeleción puede originar dos síndromes tan distintos clínicamente como el Prader-Willi y el Angelman. La patología monogénica se origina a partir de la mutación de un gen único. Las anomalías metabólicas congénitas o errores innatos del metabolismo son un grupo muy importante dentro de los millares de genopatías y muy diversos síndromes con malformaciones congénitas pueden englobarse también aquí. Los clásicos conceptos de recesivo y dominante significaban únicamente que un defecto genético se expresaba en homocigotos y heterocigotos, respectivamente. En la actualidad, estas dos categorías se pueden justificar por dos clases de proteínas: enzimáticas y estructurales. Con las primeras el organismo parece utilizar un único gen normal que en los heterocigotos sólo origina escasos efectos en el fenotipo. En el caso de las proteínas estructurales, incluso un único gen anómalo puede producir problemas. La herencia multifactorial constituye un factor etiológico de malformaciones congénitas de gran interés.
Factores ambientales
Desde que Gregg demostró en 1941 que la infección materna por el virus de la rubéola podía causar malformaciones en el producto de la gestación, se hizo evidente que el embrión está sujeto a gran cantidad y variedad de influencias ambientales, capaces de inducir efectos deletéreos en su desarrollo. La década de 1960 realiza dramáticas aportaciones en este campo con la tragedia de la talidomida, pero el auténtico potencial teratógeno de la mayoría de sustancias sigue siendo mal conocido, debido a los problemas que rodean su estudio en el ser humano: difícil extrapolación de datos obtenidos en animales de experimentación, errores epidemiológicos, variabilidad de la expresión y escaso conocimiento de la acción de los agentes teratógenos.
Un agente teratógeno se define como cualquier factor ambiental que puede originar una anomalía permanente en la estructura o función, restricción del crecimiento o muerte del embrión o feto. Las anomalías de la forma dan lugar a malformaciones mayores o menores; las anomalías de la función originan alteraciones fisiopatológicas de los distintos sistemas orgánicos, que incluyen el SNC (rendimiento intelectual). Para la mayor parte de los factores teratogénicos, el umbral de concentración que se requiere para un efecto determinado sobre el desarrollo embriofetal depende de la base genética de la madre o feto. Así, la expresión genética es influenciada y modificada por el factor ambiental en cuestión. Ejemplos bien conocidos son la embriofetopatía diabética, algunos retrasos de crecimiento intrauterino y algunos defectos del tubo neural. El teratógeno actúa produciendo muerte celular, alteración del crecimiento tisular o desviación del proceso de morfogénesis normal. Existen distintos indicadores de teratogenicidad potencial: infertilidad o pérdidas fetales previas, deficiencia de crecimiento de comienzo prenatal, alteraciones de la morfogénesis y alteraciones funcionales de los sistemas orgánicos (SNC, especialmente). La variabilidad de la expresión clínica del teratógeno depende de varias circunstancias, tales como dosis del agente, tiempo de exposición, susceptibilidad del huésped e interacciones con otros agentes ambientales. El concepto de que virtualmente cualquier sustancia es capaz de afectar adversamente al conceptus si se administra en una dosis alta es conocido en teratología como la ley de Karnofsky. La Figura 2.6.2 presenta un esquema inspirado en Moore sobre los periodos críticos del desarrollo (tiempo de exposición). La mayoría de los defectos estructurales se originan si el agente teratógeno actúa durante el periodo crítico del desarrollo. Se ha demostrado que 50 mg de talidomida administrados en el periodo crítico afectan a la mayoría de embriones, mientras que 0,5 mg del fármaco no producen efecto. Este “fármaco monstruo”, en vez de ser borrado de la farmacopea, sigue siendo propuesto para tratar afecciones tan diferentes como las aftas bucales o el dolor causálgico.
Los factores teratogénicos se clasifican en los siguientes grupos:
- infecciosos;
- factores físicos;
- drogas y agentes químicos;
- factores metabólicos y genéticos maternos.
Dentro del grupo de agentes físicos, la exposición prenatal a la radiación ionizante expone gravemente al feto a efectos teratógenos, mutágenos y carcinogénicos. La hipertermia y las fuerzas mecánicas (constricción intrauterina) son productoras de malformaciones, disrupciones y deformidades. En el grupo de factores metabólicos y genéticos maternos cabe destacar la diabetes tipo 1 materna y los hijos de madres fenilcetonúricas. La diabetes materna puede ser responsable de la secuencia de sirenomelia y de otras anomalías de las extremidades inferiores, hasta el síndrome de regresión caudal.
Distintos grados de constricción intrauterina del feto, que se hacen patentes en la última mitad de la gestación, pueden dar lugar a deformación o secuencias de deformación. La secuencia de oligohidramnios da lugar a compresión fetal y, secundariamente, a retraso del crecimiento, facies Potter, piel redundante y artrogriposis. Esta secuencia se suele relacionar con la deformación por aquinesia fetal, que incluye entidades tales como: síndrome de Pena-Shokeir tipos I y II, secuencia de restricción cutánea y síndrome de pterygium múltiple letal.
En el complejo de disrupción embriofetal, la ruptura precoz del amnios origina graves defectos de la pared corporal con extrusión de vísceras y ausencia de miembros, homolateral o contralateral, defectos del tubo neural con escoliosis, deformaciones posturales, deficiencia del crecimiento y cordón umbilical corto (limb body-wall complex LBC). Su frecuencia es de 1:10.000 nacimientos. En su patogenia se postula que la ruptura precoz del amnios (cuatro primeras semanas) inicia una serie de eventos: separación del amnios del corion, pérdida de líquido amniótico, desarrollo de bandas amnióticas y fibras mesoblásticas y compresión mecánica de cráneo, dedos, miembros y fallo en el cierre de la pared tóraco-abdominal. La ruptura tardía de amnios origina el complejo ADAM. También dentro de este grupo se incluyen disrupciones isquémicas y vasculares que originan anomalías del tipo acardia-acefalia o disrupciones de gemelos con distintas expresiones clínicas. La secuencia Potter es un ejemplo de secuencia de oligohidramnios que es secundaria a una agenesia renal, obstrucción uretral o pérdida crónica de líquido amniótico.
Estudio clínico de la patología prenatal
Blastopatías
Comprenden malformaciones debidas a trastornos en el clivaje, emigración o nidación. Su expresión clínica puede oscilar, como se adelantó, desde la muerte del producto (aborto espontáneo), pasando por anomalías cromosómicas (mosaicos cromosómicos), hasta la aparición de monstruosidades. Ejemplos conocidos son las ciclopías, monstruos sirenoides, teratomas sacrocoxígeos y, particularmente, la patología de gestaciones múltiples. En el caso de gemelos monocigotos, una división incompleta del disco embrionario da lugar a importantes alteraciones, tales como gemelos toracópagos y otras formas de gemelos siameses. Se estima que 1 de 400 embarazos de gemelos monocigotos conduce a una división incompleta. Los embarazos múltiples (triples, cuádruples, quíntuples, séxtuples y hasta séptuples) tienen incidencia creciente, relacionada con el empleo de gonadotrofinas para el tratamiento de la infertilidad en mujeres con fallos ovulatorios. La superfetación es la implantación de uno o más blastocistos en el útero que contiene ya un embrión en desarrollo. Este fenómeno es conocido en algunos mamíferos, pero no ha sido probado concluyentemente en la mujer. En patología veterinaria es conocida la blastopatía surgida en corderos: una auténtica epidemia de ciclopía, atribuida a la ingestión por ovejas preñadas del alcaloide Veratrum californium.
Los defectos del tubo neural son una patología en el límite entre blastopatía y embriopatía. El cierre del tubo neural tiene lugar entre los días 21 y 27 postconcepción. Si la anomalía tiene lugar en el neuroporo cefálico, se producirá una anencefalia o distintas variedades anatómicas de encefalocele; si se localiza en el neuroporo caudal se originarán las distintas variedades de espina bífida (oculta, meningocele o mielomeningocele). La deficiencia de ácido fólico es responsable de un número importante de casos, que se evitarán con la administración diaria de 400 μg desde 2 meses antes del embarazo.
Embriofetopatías
Embriopatía rubeólica
Descrita en 1941 por el oftalmólogo australiano Gregg, sólo el 25% de los casos de infección rubeólica gestacional da lugar a lesiones embrionarias, que incluyen desde abortos hasta RN con anomalías o sanos. Probablemente estas cifras sean muy variables de unas epidemias a otras. La vacunación previa previene, pero no siempre. El cuadro clínico principal puede sistematizarse en:
- síntomas cardinales, consistentes en catarata, microcefalia, cardiopatía congénita y sordera;
- síntomas secundarios que incluyen anomalías dentarias, bajo peso al nacimiento, pliegues cutáneos anormales y malformaciones diversas;
- síndrome ampliado, incluyendo lesiones viscerales (hepatitis, esplenitis), en los órganos hemopoyéticos (anemia, púrpura trombopénica), en aparato respiratorio (neumonitis) y esqueleto (zonas de aclaramiento metafisario, esclerosis ósea, anomalías del periostio).
Embriopatía talidomídica
Descrita por Lenz en 1961, es un trágico ejemplo de la peligrosa extrapolación de datos experimentales a la especie humana. En efecto, para producir en animales de experimentación defectos de desarrollo se precisan dosis mucho más elevadas. Las características anomalías de los “niños-talidomida” son: amelia (ausencia completa de una o varias extremidades) y meromelia (ausencia parcial de una o varias extremidades). Si la droga actúa en el momento crítico del desarrollo del miembro primario, se producirá una amelia. Otras malformaciones frecuentes afectaban a pabellones auriculares, corazón y aparato digestivo. La retirada de la talidomida (1961) coincidió con la desaparición de estos cuadros. Conviene señalar, sin embargo, que en menor escala siguen observándose RN con amelias o meromelias, no atribuibles a factores ambientales conocidos.
Virilización del feto femenino
La administración de progestágenos para evitar el aborto ha producido virilización de fetos femeninos de grado variable, en dependencia con el periodo crítico del desarrollo de los genitales externos. Los preparados más frecuentemente involucrados son los que contienen etisterona y noretisterona. La virilización conduce a un estado de ambigüedad sexual, diagnosticable en el periodo neonatal.
Embriopatía por agentes antineoplásicos
Destaca la embriopatía por metotrexato, que se caracteriza por retraso del crecimiento intrauterino, dismorfia craneofacial y anomalías de extremidades. El cuadro clínico inicial fue relacionado con la aminopterina, por lo que también se denomina síndrome de la aminopterina. El metotrexato es su metil derivado, siendo las manifestaciones clínicas comunes para ambos. La dismorfia cráneo-facial consiste en marcada hipoplasia del hueso frontal, parietales, temporales y occipital, amplias fontanelas y sinóstosis de las suturas coronaria y lamboidea. La nariz es amplia y deprimida, los ojos son prominentes con epicantus, existe micrognatia e hipoplasia de mandíbula, paladar hendido y baja implantación de pabellones auriculares. Las anomalías de extremidades más sobresalientes son: acortamiento, particularmente de antebrazos (meromelia), hipodactilia y pie equinovaro.
Embriopatía por agentes anticonvulsivantes
Existen evidencias de la interacción entre factores genéticos y susceptibilidad a determinadas drogas, como es el caso de la fenitoína. Las diferencias genéticas en la producción de metabolitos del epóxido pueden inducir alteraciones de la organogénesis. Los efectos sobre el feto son retraso de crecimiento intrauterino, anomalías congénitas, neuroblastoma y hemorragias por déficit de vitamina K. La trimetadiona origina en el 80% de las gestaciones abortos o neonatos malformados. La embriopatía por trimetadiona y parametadiona (German) presenta: anomalías de pabellones auriculares, fisuras labial y palatina, malformaciones cardiacas, urogenitales, anomalías esqueléticas, retraso del crecimiento y deficiencia mental. Los barbitúricos condicionan un patrón clínico poco característico. Desde 1982 se conoce la embriofetopatía por ácido valproico (Robert), siendo especialmente grave cuando la gestante ha recibido, además, hidantoína.
Síndrome de la warfarina
Aparece como consecuencia del tratamiento con anticoagulantes de las gestantes con prótesis valvulares cardiacas. La sintomatología consiste en: calcificaciones múltiples epifisarias, vertebrales y del cartílago nasal, asociadas a hipoplasia nasal con depresión de la raíz, obstrucción variable de vías respiratorias altas y retraso mental. Las anomalías esqueléticas son similares a las referidas para la condrodistrofia calcificante.
Síndrome alcohólico fetal
Consiste en retraso del crecimiento pre y postnatal, deficiencia mental, microcefalia, hendiduras palpebrales pequeñas, hipoplasia mandibular, labio en boca de pescado, anomalías articulares, soplo cardiaco que tiende a desaparecer con la edad y macrodactilia del primer dedo del pie. La causa directa radica en el etanol o uno de sus derivados, concretamente acetaldehído, con acción teratogénica directa. Esta toxicidad se vería reforzada por las perturbaciones enzimáticas propias del etilismo crónico. En algunos casos han sido descritas anomalías cromosómicas.
Disrupciones y deformidades
Complejo de disrupción por bandas amnióticas
La manifestación más importante se ha agrupado con el acróstico de complejo ADAM (deformidad amniótica, adherencias y mutilaciones). Pueden encontrarse: labio leporino, hendiduras faciales, hidrocefalia, microcefalia, encefalocele anterior, fisura palpebral colobomatosa, microftalmía, anoftalmía y opacidad corneal, onfalocele y gastrosquisis, pero destacan por su frecuencia las anomalías en extremidades inferiores, que oscilan desde amputaciones a constricciones en forma de anillo.
Deformidades
Son alteraciones congénitas de la forma o estructura de una región previamente desarrollada con normalidad, a diferencia de las malformaciones, que son errores primarios de la morfogénesis. La mayor parte afectan al sistema músculo-esquelético, por lo que se llaman deformidades posturales congénitas. Se unen frecuentemente con diversas malformaciones (7,6%). Incluyen los cuadros ya expuestos: síndrome letal del pterygium múltiple, secuencia de hipoquinesia/aquinesia fetal, etc. Aparecen más frecuentemente en hijos de primigrávidas con oligohidramnios, presentación de nalgas, hipertensión materna y en RN de bajo peso.
Fetopatías
El periodo fetal se caracteriza por el rápido crecimiento corporal y por la diferenciación de órganos y tejidos ya formados previamente. Este ritmo rápido es especialmente intenso entre las 12 y 20 semanas de la gestación. Durante las últimas semanas de vida intrauterina se asiste a un incremento máximo del peso del feto, que es menos vulnerable a la acción de los diferentes agentes teratógenos, pero éstos pueden interferir en el desarrollo del SNC, así como inducir la aparición de perturbaciones funcionales o morfológicas mínimas.
Las causas de fetopatía son diversas, incluyendo:
- Enfermedades fetales producidas por fármacos o agentes químicos, como mercuriales orgánicos, hidrocarburos clorados, quinina y otros;
- Fetopatías infecciosas, ya que el 2% de los fetos se infectan intraútero y a las infecciones antiguas se añaden otras nuevas;
- Metabolopatías (diabetes materna, malnutrición);
- Inmunopatías, tanto por isoinmunización, como por autoinmunidad;
- Enfermedades maternas diversas, como hemopatías, nefropatías, toxemia, hipertensión o endocrinopatías;
- Drogas, tanto menores, como tabaco y alcohol, como “blandas” y “duras”, causantes de malnutrición fetal, malformaciones y síndromes de abstinencia;
- Otras afecciones, incluidas las radiaciones, las hemorragias, las anomalías posturales y los traumatismos físicos o psíquicos.
Como ya es conocido, en la práctica no siempre es posible distinguir netamente entre embriopatía o fetopatía y sus respectivas consecuencias clínicas.
Fetopatías de origen infeccioso
La infección intrauterina puede originar distintas consecuencias: la reabsorción del producto durante el periodo embrionario, aborto, mortinato, malformación, retraso del crecimiento intrauterino, prematuridad y secuelas de infección postnatal crónica. Aunque el feto puede responder a la agresión infecciosa con producción de IgM, sus tasas son insuficientes para evitar la enfermedad fetal. La respuesta de IgM fetal, sin embargo, puede ser utilizada en el periodo neonatal como indicadora de enfermedad infecciosa prenatal.
Fetopatía diabética
Estos neonatos son justamente considerados como “RN de riesgo elevado”, ya que sin tratamiento correcto presentan una mortalidad perinatal que oscila del 10 al 20%. Su incidencia es del 1% de embarazos, correspondiendo un 0,6% a diabéticas gestacionales.
Generalmente serán neonatos de peso elevado, pero en la diabetes de los grupos D a F suele existir una insuficiencia placentaria que dará lugar a niños “normales” o a RN de bajo peso. En este sentido, White sugirió una clasificación basada en la gravedad de la diabetes de la gestante.
Interesan los factores de mal pronóstico para el feto:
- pielonefritis;
- precoma o acidosis grave;
- toxemia; y
- “madres negligentes”, que no han seguido durante el embarazo el régimen recomendado.
El feto es sometido intraútero al efecto adverso del nivel, generalmente elevado, de glucosa en sangre materna, lo que determina un hiperinsulinismo secundario. Éste producirá disminución de la calidad del surfactante pulmonar y del fosfoglicerol y un aumento de peso, ya que el crecimiento a esta edad depende de la insulina. El hijo de madre diabética es más obeso que edematoso y el acúmulo de grasa es favorecido por el hiperinsulinismo. Actualmente se admite la existencia conjunta de hiperinsulinismo y depleción de epinefrina, lo que permite explicar la macrosomía y las alteraciones metabólicas. Además, la madre con hiperglucemia posee elevadas cantidades de glicohemoglobina (HbA1c), que comporta una desviación a la izquierda de la curva de disociación de la hemoglobina, eliminándose menos oxígeno a nivel tisular. Esta hipoxemia relativa crónica puede ser la causa de la poliglobulia en el hijo de diabética. El parto prematuro, la insulina y antidiabéticos orales, así como factores genéticos asociados a la diabetes materna, pueden intervenir en la patogenia.
Hay que destacar la hipoglucemia, que aparece en más del 50% de los niños y alcanza su máxima intensidad hacia la hora y media o dos horas del nacimiento. La intensidad, que puede llegar a cero, depende del grado de hiperinsulinismo secundario, relacionado con la gravedad de la diabetes materna. Cursa con manifestaciones clínicas variables: convulsiones, crisis de apnea, hipotonía, letargia, rechazo del alimento, respiración jadeante e irregular, llanto débil, cianosis y nistagmus rotatorio, pero debe tenerse en cuenta que la hipoglucemia intensa a veces cursa de un modo asintomático, siendo capaz de producir daño cerebral irreparable. La hipocalcemia suele manifestarse hacia el segundo o tercer día de vida. En su desencadenamiento intervienen varios factores, si bien el más importante es el hipoparatiroidismo transitorio, derivado del hiperparatiroidismo materno. Las manifestaciones clínicas son también diversas: convulsiones, sobresaltos, hiperexcitabilidad, hipertonía muscular, crisis de apnea y cianosis, edemas, distensión abdominal y vómitos. Debe citarse la tendencia a la hiperbilirrubinemia, a la eritroblastosis, a la acidosis de tipo mixto, a la poliglobulia (que puede predisponer al padecimiento de trombosis venosa renal) y la presencia de malformaciones congénitas asociadas (presentes en el 6,4% de los casos), especialmente cardiacas (defectos septales ventriculares, transposición de los grandes vasos), anomalías aórticas y espinales (hemivértebras, síndrome de regresión caudal). La enfermedad de la membrana hialina es frecuente, en parte por su prematuridad y también por la defectuosa síntesis de surfactante (no en cantidad, sino en calidad). En este caso la relación L/E puede ser errónea para evaluar la madurez fetal. Las infecciones son frecuentes por su inmadurez y la alta incidencia en la madre de infecciones urinarias con o sin pielonefritis. También puede existir miocardosis o miocardiopatía hipertrófica.
El diagnóstico se basa en los antecedentes, el aspecto clínico del niño, la aparición de complicaciones y, si es posible, la dosificación de insulina fetal que estará elevada (superior a 10 μU/mL). Otros exámenes complementarios a solicitar son glucemia repetida (1/2, 1, 2, 6, 12, 24, 36, 48 h, y después diaria durante los tres o cuatro primeros días), calcemia y hematocrito. Debe efectuarse el diagnóstico diferencial con las restantes causas de RN de peso elevado.
Las embarazadas diabéticas deben ser tratadas únicamente con insulina durante el embarazo. Es muy importante mantener un nivel constante en la glucemia materna, para evitar los graves efectos, hasta la muerte intraútero, de las hipoglucemias fetales consecutivas a respuestas insulínicas. Si se presume un feto macrosómico se practicará cesárea electiva.
En las diabéticas insulinodependientes (grupos B a F), cuando se demuestra que el feto es maduro se inducirá el parto, a no ser que la existencia de macrosomía aconseje cesárea.
Pauta terapéutica del hijo de diabética
Consta de:
- Cuidados generales. Colocar al RN en incubadora y alimentar precozmente. Siempre que sea posible se inicia la alimentación oral a las 3-6 horas de vida;
- Vigilancia y corrección de las alteraciones metabólicas:
- Glucemia. Se administrará glucosa EV desde el momento del nacimiento, utilizando suero glucosado al 10% a dosis de 75 mL/kg y día, añadiendo de 3 a 6 mEq/kg de cloro y sodio a partir del 3 er día de perfusión. Cuando la glucemia es inferior a 20 mg/dL y el niño presenta clínica, se aumenta la concentración de glucosa en la perfusión hasta el 15%. No conviene la administración EV de glucosa hipertónica, por la grave hipoglucemia que puede originarse como rebote. La supresión de la glucosa debe ser lenta.
- Calcemia. Siempre que sea inferior a 7 mg/dL debe corregirse. Se emplea gluconato cálcico al 10% a dosis de 1-2 mL/kg de peso por vía EV y administración lenta si hay clínica, o por vía oral cuando ésta no existe, repitiendo mientras persiste la hipocalcemia.
- Complicaciones. Es obligado detectar su posible aparición para aplicar el correspondiente tratamiento de la ictericia, poliglobulia, infección y distrés respiratorio.
Evolución
El peso y la talla tenderán a normalizarse, aunque en ocasiones el niño permanecerá en percentiles elevados. El desarrollo mental se efectuará normalmente, si el niño no ha sufrido insultos neurológicos intraútero (cetoacidosis materna, hipoglucemia fetal) o tras el parto (traumatismo obstétrico, hipoglucemia). Sobre el posterior desarrollo de diabetes infantil por agotamiento intraútero del páncreas no se ha llegado a un acuerdo unánime.
Hijo de madre drogadicta
Si las madres consumen drogas (heroína, metadona, fármacos, alcohol), los fetos deben ser considerados como de “alto riesgo”. La heroína frena el crecimiento fetal por disminuir el número total de células y acortar la gestación. Estos fetos tienen, además, más probabilidades de adquirir infecciones intrauterinas.
Los hijos de madres heroinómanas presentan retraso del crecimiento (42%) y microcefalia (30%). El síndrome fetal de “supresión” o “abstinencia” durante el periodo neonatal es frecuente: hiperactividad generalizada, irritabilidad, sueño alterado, regurgitaciones, pobre ganancia de peso, distermia e hiperhidrosis. Es necesario descartar SIDA y enfermedades de transmisión sexual.
Enfermedad de Minamata
Descrita por Matsumoto (1965) en RN de madres alimentadas durante la gestación con pescados que contenían niveles anormalmente elevados de mercuriales orgánicos, su denominación se debe a la localización geográfica inicial en la bahía japonesa de Minamata. Con posterioridad esta fetopatía ha sido comprobada en otros países. Incluye una parálisis cerebral congénita con retraso mental y alteraciones motoras, asociada a ceguera y sordera y trastornos morfológicos mínimos. Los síntomas neurológicos (piramidales y extrapiramidales) son de carácter estacionario, si bien parecen más ostensibles al final del primer año de vida extrauterina. Se diagnostica con los datos epidemiológicos, familiares y clínicos, confirmándolo las cifras elevadas de mercurio en cabello, sangre y orina. Su tratamiento es sintomático y rehabilitador.
Fetopatía por bifenoles policlorados
Los compuestos organoclorados son contaminantes ambientales de amplio uso. Entre ellos se encuentran DDT, BHC y PCB (bifenol policlorado), que pueden afectar a los alimentos, incluida la leche de mujer. Los hidrocarburos clorados persisten en el ambiente, se almacenan en el tejido adiposo de los mamíferos y son metabolizados lentamente. Por el contrario, otros insecticidas peligrosos, como los fosfatos orgánicos y los carbamatos, que tienen acción anticolinesterasa, son rápidamente metabolizados y eliminados por los tejidos de los mamíferos, permaneciendo durante poco tiempo en el ambiente.
Sintomatología: erupción tipo acné en la piel de la cara, cuello y tórax, pigmentación de uñas o encías, trastornos visuales, hiperemia conjuntival y de otras mucosas, caries dental, cefalalgias, dolor abdominal y pérdida de peso. También son frecuentes en niños las infecciones del aparato respiratorio, retraso del crecimiento y anomalías del sistema esquelético. El feto presenta peso bajo para la edad gestacional, pigmentación de la piel bronceada oscura con descamación, hipertrofia gingival con dientes congénitos y calcificaciones craneanas. Otros síntomas: anillo corneal coloreado, fontanelas y suturas amplias, exoftalmos, secreción conjuntival, edema palpebral, talón prominente, hepatomegalia dura y fiebre variable.
Diagnóstico: faltan malformaciones. Elevación de las transaminasas GOT y de la fosfatasa alcalina; presencia sérica de una betalipoproteína anormal, expresión de la afectación hepática.
Otras fetopatías
Los trastornos hemorrágicos antepartum (maternos y fetales), la enfermedad hemolítica fetal por incompatibilidad Rh, el RN de bajo peso por retraso del crecimiento intrauterino y otras afecciones son tratados en otras lecciones.
El uso de inhibidores del sistema renina-angiotensina en embarazadas afectas de hipertensión, fallo cardiaco, isquemia coronaria o nefropatía durante el segundo y tercer trimestres de la gestación se asocia con un patrón de defectos atribuidos a displasia tubular renal fetal. El cuadro clínico se denomina fetopatía ACEI y se caracteriza por oligohidramnios y compresión fetal que secundariamente origina retraso de crecimiento intrauterino, deformaciones craneofaciales, contracturas de las extremidades inferiores e hipoplasia pulmonar.
Los inhibidores de la recaptación selectiva de serotonina ampliamente utilizados como tratamiento de choque para prevenir cuadros de ansiedad o depresión en la gestante pueden ser teratógenos potenciales. Se han descrito mayor riesgo de cardiopatías congénitas y riñón quístico.
Las benzodiacepinas se han asociado a una mayor incidencia de fisura palatina, cuando actúan sobre el embrión/feto durante el primer trimestre de la gestación.
Las estatinas, un grupo de fármacos hipolipidémicos que inhiben la enzima 3-hidroxi-3-metilglutaril coenzima A reductasa, limitando la síntesis de colesterol, pueden actuar sobre el embrión o feto originando un síndrome de Smith-Lemli-Opitz, un síndrome Child (hemidisplasia congénita con eritrodermia ictiosiforme y defectos de miembros) o una condrodisplasia punctata tipo 2.
Están sujetos a controversia la acción teratógena del tabaco y el uso de las tecnologías de reproducción asistida.