Trastornos hemorrágicos y trombóticos en el recién nacido
Los trastornos hemorrágicos del periodo neonatal afectan aproximadamente al 1% de los RN, aunque sólo en la mitad de ellos tendrán importancia clínica. Este porcentaje es más elevado (10-20%) en aquellos RN ingresados en las unidades de cuidados intensivos neonatales, encontrándose hemorragias o trombosis en el 10% de las necropsias neonatales.
Coagulación sanguínea en el recién nacido
Los factores de la coagulación no atraviesan la barrera placentaria.
Los sistemas hemostático y fibrinolítico se desarrollan simultáneamente a las 10-11 semanas de gestación. La sangre en circunstancias fisiológicas se mantiene en una fase fluida. En respuesta a una lesión de la pared vascular, las plaquetas, las proteínas de la coagulación y los vasos lesionados contribuyen a la formación de un tapón hemostático para evitar el sangrado excesivo. La sobrerreactividad patológica de este proceso puede ocasionar una trombosis.
La finalidad de las proteínas de la coagulación es generar trombina desde la protrombina. La trombina es una serina proteasa con potente función coagulante. La trombina separa los fibrinopéptidos A (FPA) y B (FPB) del fibrinógeno, produciendo la fibrina, y activa los factores V (Va), VIII (VIIIa) y IX (IXa), con lo cual facilita su propia generación; también promueve las uniones cruzadas entre la fibrina por activación del factor XIII (XIIIa) y es un potente activador de las plaquetas. Se han establecido valores de referencia para las concentraciones plasmáticas de muchos factores de la coagulación que varían según la edad gestacional, siendo variable la expresión del ARNm de las diversas proteínas de la coagulación en el periodo neonatal y normalizándose hacia los 6 meses de edad. Los niveles de fibrinógeno, factores V y VIII en ambos grupos son similares a los encontrados en niños mayores y adultos, estando disminuidas las concentraciones de los factores que requieren vitamina K para su funcionamiento normal (II, VII, IX y X) y esta disminución es más acentuada en el pretérmino. Los resultados sobre la actividad del factor XIII son contradictorios por los distintos métodos de medición utilizados, si bien parece estar disminuida en un 50% respecto a los observados en niños mayores, presentando valores más altos los neonatos con síndrome de dificultad respiratoria. Los “factores de contacto” que intervienen en la activación inicial del sistema intrínseco también están disminuidos en más del 50% respecto a los valores del adulto (factores XI y XII, precalicreína y cininógeno de elevado peso molecular).
Los inhibidores directos de la coagulación son la antitrombina (AT), la α2-macroglobulina (α2-M) y el cofactor II de la heparina (HCII). Al nacer, las concentraciones plasmáticas de AT y HCII son el 50% de las del adulto, mientras que los niveles de α2-M están elevados. Los inhibidores indirectos de la trombina son la proteína C, la proteína S y el inhibidor de la vía del factor tisular (TFPI), cuyas concentraciones están reducidas al 60% de las del adulto. Cuando se genera in vivo, la trombina se une a un receptor de la célula endotelial, la trombomodulina (TM), y de esta forma puede no prolongarse la fragmentación del fibrinógeno y la activación de los factores V y VIII. La trombina puede activar la proteína C a su forma activa (APC), que es una serina proteasa dependiente de la vitamina K que inactiva enzimáticamente los factores Va y VIIIa, lo que se ve facilitado por la proteína S. El TFPI forma un complejo con el factor IXa, que inhibe el VIIIa y por ello limita la generación de la trombina. La concentración de fibrinógeno al nacer es similar a la del adulto, pero el fibrinógeno “fetal” tiene un contenido más alto de ácido siálico, lo que explicaría que se prolongue el “tiempo de trombina” (TT).
La mayoría de los estudios sugieren que la actividad fibrinolítica global de la sangre neonatal está activada al nacer en comparación con la de la madre o adultos sanos, normalizándose 6 horas después del nacimiento.
Una vez se ha formado el tapón de fibrina es modificado por el sistema fibrinolítico, para ello el plasminógeno es convertido en plasmina por el factor tisular activador del plasminógeno (tPA) y, en menor grado, por los componentes del sistema de contacto. La plasmina es una serina proteasa que fragmenta la fibrina en pasos secuenciales, ocasionando los productos de degradación de la fibrina (PDF) y el fragmento específico de la fibrina, el dímero D. La plasmina es inhibida primariamente por la α2M y secundariamente por los inhibidores de los factores activadores del plasminógeno (PAI), de los que el más importante es el PAI-1.
La actividad fibrinolítica del plasma de los pretérmino suele ser menor que la de los RN a término, y es muy baja en los neonatos con síndrome de dificultad respiratoria. Los niveles de plasminógeno suelen ser el 50% de los observados en adultos normales y los de la α2-M son el 80% de los del adulto, estando aumentados, sin embargo, los niveles del factor tisular activador del plasminógeno (tPA) y del PAI-1 (siendo similares a los del adulto en sangre del cordón).
En los RN sanos las diferencias entre los factores procoagulantes, anticoagulantes y fibrinolíticos, son fisiológicas y no dan lugar a hemorragias ni trombosis clínicas. Pero en los RN enfermos las concentraciones más bajas de muchos factores los sitúan en riesgo de desequilibrio entre factores procoagulantes y anticoagulantes.
La cara externa de las plaquetas contiene varias glicoproteínas (GPs) que se unen a proteínas específicas adhesivas para facilitar las interacciones plaqueta-plaqueta (agregación) y plaqueta-superficie (adhesión). Bajo el glicocálix hay una bicapa fosfolipídica que proporciona una superficie procoagulante tras la activación plaquetaria y un sistema interno de la membrana que participa en la secreción de los gránulos plaquetarios. Las plaquetas contienen dos tipos de gránulos: los cuerpos densos y los αgránulos. Los cuerpos densos contienen sustancias que promueven la agregación plaquetaria tales como ADP, serotonina y calcio. Los α-gránulos contienen sustancias con diversas funciones: proteínas adhesivas, como la tromboespondina, factores de crecimiento, como el factor de crecimiento derivado de las plaquetas (PDGF), el factor de crecimiento tisular-β (TGF-β), el factor de crecimiento fibroblástico (FGF), el factor plaquetario 4 (PF4), que interfiere en la interacción heparina-AT, varias proteínas de la coagulación y la β-tromboglobulina, que es un marcador de la activación plaquetaria. En los prematuros < 1.500 g hay una cierta hiporreactividad de las plaquetas durante los primeros 3-4 días de vida.
Cuando se lesionan las células endoteliales, las plaquetas se adhieren a las capas subendoteliales, cambiando de forma y diseminándose sobre la superficie y uniéndose entre sí. La adhesión de las plaquetas inicia la secreción de los gránulos plaquetarios y estas sustancias promueven la adhesión y la formación de agregados plaquetarios. Las plaquetas también participan en la coagulación, proporcionando una superficie lipoproteica sobre la cual pueden formarse los complejos de factores solubles de la coagulación, lo que conduce a la generación de la trombina y la formación de la fibrina. El proceso de adhesión plaquetaria es mediado por una proteína específica de la membrana (GP Ib), que sirve de lugar de unión al factor de von Willebrand (FvW) que es secretado por las células endoteliales y las plaquetas. Tras la activación de las plaquetas se forman complejos de GP IIb y GP IIIa con fibrinógeno y, en menor cuantía, con FvW y fibronectina, lo que facilita la agregación plaquetaria.
El endotelio juega un papel complejo en la hemostasia, previniendo las complicaciones trombóticas en circunstancias normales y promoviendo la formación de fibrina cuando se lesiona. Las propiedades anticoagulantes de las células endoteliales están mediadas por metabolitos del ácido araquidónico producidos por las vías de la lipooxigenasa y la ciclooxigenasa. La prostaciclina (PGI2), producida por la vía de la lipooxigenasa, es un potente vasodilatador que inhibe la agregación plaquetaria y facilita la fibrinólisis. Los proteinglicanos de la superficie de las células endoteliales, como el heparán sulfato, promueven la neutralización de la trombina por la AT y otras serina-proteasas. La producción endotelial de óxido nítrico (ON) modula el tono vascular en el pulmón fetal y el postnatal, siendo también el ON un potente inhibidor de la activación plaquetaria.
El receptor endotelial TM, al que se une la trombina, acelera la activación de la proteína C (APC). La APC, en presencia de la proteína S, inactiva los factores Va y VIIIa. Las células endoteliales producen también muchos de los componentes del sistema fibrinolítico tales como el tPA, PAI-1 y la urocinasa.
Etiología y patogenia
Diferentes trastornos pueden ser los causantes de las hemorragias neonatales:
- Acentuación del déficit fisiológico transitorio de los factores de la coagulación vitamina K dependientes.
- Alteraciones transitorias del mecanismo de la coagulación, por procesos patológicos neonatales asociados (coagulación intravascular diseminada, hepatopatías).
- Anomalías permanentes hereditarias del mecanismo de la coagulación (hemofilia y otros defectos congénitos de los factores de la coagulación).
- Alteraciones plaquetarias cuantitativas o cualitativas.
- Anomalías vasculares: telangiectasias, fragilidad capilar, más acentuada en el prematuro y con incremento patológico en los estados de anoxia, choque e infecciones.
- Traumatismos aislados o asociados con algunos de los factores mencionados anteriormente. La alteración de los distintos factores que intervienen en la hemostasia puede conducir al estado hemorrágico.
Enfermedad hemorrágica del recién nacido
En sentido estricto, es un trastorno hemorrágico propio de los primeros días de vida y producido por una deficiencia de vitamina K, que se caracteriza por déficit de la actividad de la protrombina, proconvertina, factor Christmas y factor de Stuart-Power (factores II, VII, IX y X). Los pacientes con déficit de vitamina K presentan una notable discrepancia entre la actividad de la protrombina medida en los análisis estándar de coagulación y la hallada en los inmunoanálisis de la protrombina o en el análisis de la actividad del factor II con veneno del Echis carinatus, desapareciendo esta discordancia tras la administración de vitamina K. Tales observaciones clínicas sugieren que en los estados de déficit de vitamina K el hígado sintetiza una molécula semejante a la protrombina, la cual presenta unas ligeras diferencias estructurales en comparación con la protrombina normal. Estas diferencias producirían un importante descenso en la actividad de la protrombina. Se sabe actualmente que la vitamina K no es necesaria para la síntesis de los factores II (protrombina), VII, IX y X, pero sí para la conversión de las proteínas precursoras en proteínas con actividad coagulante. La vitamina K participa en la carboxilación de los átomos de carbono gamma-metileno de los residuos de ácido glutámico existentes en el N-amino terminal de las proteínas precursoras, dando lugar a residuos de ácido gammacarboxiglutámico. La adición de este segundo grupo carboxilo a los residuos de ácido glutámico requiere la presencia de una proteína hidroxilasa, la forma de hidroquinona de la vitamina K, oxígeno molecular y anhídrido carbónico. Este cambio estructural, relativamente mínimo, aumenta la carga negativa de la molécula de protrombina, permitiendo la ligazón de los iones calcio. Estos iones de calcio sirven entonces de puente para unir la molécula de protrombina a los fosfolípidos de las membranas. De esta forma las proteínas de la coagulación pueden estar en estrecha proximidad una con la otra, facilitándose la proteólisis. En ausencia de la carboxilación dependiente de la vitamina K en los microsomas hepáticos, los factores II, VII, IX y X sintetizados en el hígado no se unen normalmente al calcio, reduciéndose notablemente su actividad coagulante. A la protrombina anormal, parcialmente carboxilada, que aparece en el déficit de vitamina K se la denomina PIVKAII (PIVKA = proteína inducida por ausencia de vitamina K), existiendo una anormal relación entre la actividad de coagulación y la cuantificación del factor II antigénico (< 0,7). Es conocido asimismo que la vitamina K participa en la carboxilación de otras proteínas a nivel del hueso (osteocalcina), riñón, bazo, placenta, páncreas y pulmón.
Formas de presentación
La forma clásica es conocida como enfermedad hemorrágica del RN y tiene lugar durante la primera semana de vida. Está presente en el 0,01-1,7% de RN a término, sanos, alimentados con lactancia materna y que no han recibido vitamina K, manifestándose clínicamente en 1 de cada 200. Las manifestaciones externas más comunes son las hemorragias gastrointestinales, umbilicales, nasales, en cuero cabelludo y equimosis generalizadas. El primer signo puede ser el sangrado prolongado en punciones efectuadas para determinaciones analíticas. También pueden producirse en órganos internos (hemorragia intracraneal, suprarrenal). Está relacionada con escasa transferencia placentaria de vitamina K, bajo contenido de vitamina K en la leche materna y falta de flora bacteriana intestinal, que es necesaria para la síntesis de la vitamina K. Ésta pertenece al grupo de las vitaminas liposolubles y, pasada la edad neonatal, su carencia es rara, no sólo por su abundancia en los alimentos (filoquinona o vitamina K1, contenida principalmente en los vegetales de hoja verde) sino por ser sintetizada por los microorganismos intestinales (menaquinona o vitamina K2 ). Además, en el neonato hay un reconocido estado de inmadurez hepática, más marcado en los RN pretérmino, que podría agravarse por la anoxia y la mala alimentación inicial, todo lo cual contribuye a la aparición de una hipoprotrombinemia fisiológica que dará lugar a un alargamiento del tiempo de protrombina.
La alimentación del RN tiene un papel importante sobre el proceso de la coagulación: por un lado, la alimentación temprana tiene efectos beneficiosos sobre los factores de la coagulación dependientes de la vitamina K; por otro lado, hay que tener en cuenta que la leche humana sólo contiene 1,5-2,1 μg/L de vitamina K frente a 6 μg/L de la leche de vaca y 11,5 μg/L las fórmulas adaptadas, facilitando el desarrollo de una flora bacteriana intestinal sintetizadora de vitamina K2 (menaquinona), siendo raro que aparezca déficit de vitamina K en los niños alimentados con fórmula.
Una segunda forma de presentación es la forma precoz, que es muy rara, apareciendo las alteraciones hemorrágicas en las primeras 24 horas de vida y está relacionada con la utilización materna de medicaciones (warfarina, fenobarbital, fenitoína, rifampicina, isoniazida) que interfieren con el depósito o la función de la vitamina K. La profilaxis con vitamina K puede ser insuficiente para prevenir la aparición de la enfermedad precoz hemorrágica del RN.
La tercera forma es la forma tardía, que se manifiesta entre las 2 y las 12 semanas de vida. Estos niños se caracterizan por no haber recibido vitamina K (o profilaxis inadecuada), seguir lactancia materna, posiblemente haber padecido una diarrea o haber recibido antibioticoterapia o padecer trastornos que comprometen la absorción de vitamina K (ictericia colestásica, fibrosis quística, atresia de vías biliares, déficit de α1 -antitripsina). Su incidencia se estima en 4-10 casos/100.000 RN, pero tiene un peor pronóstico porque la mayoría de los casos cursan con hemorragia intracraneal.
Si se sospecha uno de estos cuadros clínicos se solicitarán unas pruebas de coagulación, objetivándose un alargamiento en el TP e incluso del TTPA si es una forma grave.
Profilaxis de la enfermedad hemorrágica del RN
Se realiza con una inyección IM única de 1 mg de vitamina K1, que puede reducirse a 0,5 mg en los RN con bajo peso. Algunos autores preconizan la vía oral, dado que la inyección IM fue relacionada, en algunos estudios epidemiológicos, con la predisposición a padecer cáncer, pero ello no se ha confirmado en otros trabajos que sugieren un origen prenatal del cáncer y concluyen que no existe asociación entre profilaxis parenteral de vitamina K y neoplasias en la infancia. Por otra parte, se ha observado la existencia de formas tardías de enfermedad hemorrágica en 1,2-6,4 casos/100.000 RN que recibieron profilaxis oral completa o incompleta.
Los requerimientos diarios de vitamina K son de 1-5 μg/kg, pero para la profilaxis oral se administra el primer día una dosis de 2 mg de vitamina K, debiéndose repetir a los 7 días y, en los niños alimentados al pecho, una tercera dosis a las 4 semanas de vida. Pero la administración parenteral siempre es más segura y eficaz que la oral, en especial para la prevención de las formas tardías. Carece de utilidad la administración materna antenatal de vitamina K1, que no reduce tampoco la incidencia ni la gravedad de la hemorragia intracraneal en los pretérmino pero, cuando la madre recibe tratamiento anticonvulsivante, sí es aconsejable que reciba 5 mg de vitamina K oral al día durante el último trimestre. También es conveniente su administración a la madre lactante, ya que la vitamina K pasa a la secreción láctea.
Tratamiento
Consiste en la administración intramuscular (IM) o, mejor, endovenosa (EV), de 2 mg de vitamina K1, pudiéndose demostrar aumento de la actividad de los factores de la coagulación vitamina Kdependientes en dos a cuatro horas, con acortamiento del TP. Las anomalías de la coagulación quedan corregidas a las 24 horas de la administración de vitamina K, si bien en el pretérmino o si existe una hemorragia activa puede ser necesaria también la administración de plasma fresco o congelado (10 mL/kg).
Coagulación intravascular diseminada
Es un proceso fisiopatológico adquirido caracterizado por el consumo intravascular de las plaquetas y los factores plasmáticos de la coagulación (factores II, V, VIII, XIII y fibrinógeno), con activación de los sistemas de la coagulación y la fibrinólisis. Esta coagulación diseminada (CID) en el interior de los vasos sanguíneos produce depósitos de trombos de fibrina y un estado hemorrágico cuando la rápida utilización de las plaquetas y de los factores de la coagulación origina niveles inadecuados para mantener la hemostasia. Los acúmulos de fibrina en la microcirculación producen daño mecánico de los eritrocitos, con fragmentación de los mismos y anemia microangiopática. Es un trastorno frecuente en el RN gravemente enfermo, sobre todo en prematuros, y es responsable de la mayoría de las hemorragias clínicamente evidentes en estos niños con asfixia grave, distrés respiratorio idiopático, sepsis, hipotensión, anemia aguda, toxemia materna, aspiración de meconio, hipotermia. Estas afecciones motivan una activación intrínseca (daño endotelial) o extrínseca (daño tisular) de la coagulación, con paso de la protrombina a trombina y formación de múltiples trombos intravasculares. El proceso es más exagerado en los prematuros por la limitada capacidad de su sistema reticuloendotelial para eliminar de la circulación los factores activados de la coagulación y por el déficit de antitrombina.
Manifestaciones clínicas
Son muy variables y dependen en parte del proceso patológico asociado y de la intensidad del trastorno de la coagulación. En los casos típicos se observan petequias y sangrado persistente en los sitios de punción, pudiéndose producir hemorragias pulmonares, cerebrales, hemorragia por el muñón umbilical y orificios corporales, púrpura, equimosis y trombosis de vasos centrales o periféricos con necrosis tisulares.
Diagnóstico de la CID
Se realiza en todo RN gravemente enfermo con grados variables de plaquetopenia, prolongación de los tiempos de protrombina (TP), tromboplastina parcial activada (TTPA) y de trombina (TT), así como disminución del fibrinógeno y aumento, aunque no siempre presente, de los productos de degradación de la fibrina o del dímero D.
Tratamiento
Consiste ante todo en el tratamiento del proceso que ha sido el desencadenante de la CID, sepsis, hipoxia, acidosis, así como del soporte de medidas generales, como oxigenación, circulación y tensión arterial. Se focalizará el manejo agudo de la hemostasia para minimizar el riesgo de hemorragias, mediante:
- la administración de vitamina K, 1 mg;
- plasma fresco congelado (proporciona todas las proteínas de la coagulación), 10-20 mL/kg/12 horas;
- plaquetas (1 unidad de plaquetas por cada 3-5 kg de peso) si el recuento plaquetario es inferior a 50.000/mm³ o < 100.000/mm³ si existe sangrado activo, ventilación mecánica o intervención quirúrgica reciente;
- si los niveles de fibrinógeno son inferiores a 1 g/L se administrará crioprecipitado (contiene las más altas concentraciones de fibrinógeno, factores VIII, vWF, XIII, y fibronectina);
- derivados de plasma humano inactivados para virus conteniendo antitrombina y proteína C, en casos de niveles muy bajos. Otra opción sería inhibir la activación de la coagulación con dosis profilácticas de heparina sódica (5-10 U/kg/h) aunque los ensayos clínicos con anticoagulantes en neonatos no han sido concluyentes y el riesgo de sangrado podría aumentar.
La trombosis no es muy frecuente aunque podría tratarse con heparina sódica en casos de riesgo vital si la trombosis es muy extensa; en este caso la dosis sería un bolo de 30 U/kg, seguido de perfusión de 10-15 U/kg/h.
Trastornos hemorrágicos hereditarios en el neonato
Aunque la mayoría de los niños con deficiencias congénitas de los factores de la coagulación no presentan trastornos durante las primeras semanas de vida, a pesar de sus tasas bajas desde el nacimiento al no atravesar la placenta, todas estas alteraciones pueden manifestarse por hemorragias en el periodo neonatal, tratándose en dichos casos de deficiencias graves. La hemofilia clásica o deficiencia del factor VIII (actividad < 1%) es el trastorno de coagulación congénito más común de presentación neonatal; afecta aproximadamente a uno de cada 10.000 varones, siendo menor la incidencia del déficit del factor IX (1/50.000 varones). El número de personas con enfermedad de von Willebrand excede al de los hemofílicos clásicos.
Los neonatos con coagulopatías hereditarias pueden presentar hematomas (perirrenal, esplénico, cuero cabelludo), hemorragia después de la circuncisión, sangrado por el muñón umbilical, hemorragia intracraneal, hemorragias digestivas o sangrado tras punciones en talón u otras localizaciones. Únicamente mostrará síntomas en las primeras semanas de vida un 5-35% de los hemofílicos (déficit de factores VIII o IX), un 67% de los pacientes con déficit del factor I, un 100% si se trata de déficit de factor XIII (hemorragia umbilical) y un 7% en la deficiencia de otros factores de la coagulación. Estos trastornos son relativamente raros y sólo deben considerarse después de excluir las causas más comunes de hemorragias neonatales. Los déficit aislados de los factores II, VII, V y XI son autosómicos recesivos y poco frecuentes en el periodo neonatal, siendo más habituales los déficit combinados de los factores II, VII, IX y X, o de V y VIII.
Diagnóstico
La hemofilia se tiene que sospechar ante cualquier sangrado importante en un RN sano, con historia familiar o sin ella, con un alargamiento aislado del TPT. Para hacer el diagnóstico hay que solicitar las pruebas que se exponen en el capítulo 18.12, pudiéndose hacer el diagnóstico prenatal de la mayoría de estos trastornos y la genoterapia está cada vez más próxima.
Tratamiento
En los trastornos congénitos de la coagulación deberá efectuarse (tras la extracción de sangre para los oportunos estudios de la coagulación) con la administración de 10-20 mL/kg de plasma fresco congelado, que se puede repetir cada 12 horas hasta controlar la hemorragia. Cuando no esté bien identificado el trastorno se pueden usar crioprecipitados de plasma (4 unidades/kg para el déficit del factor I, 5 unidades/kg para el déficit del factor VIII, 1 unidad por cada 10 kg de peso para el déficit del factor XIII). Para las hemorragias con riesgo vital debidas a hemofilia A o si ya el neonato va a ser sometido a cirugía mayor se administrará concentrado de factor VIII recombinante (50-75 U/kg, seguido de 2-4 U/kg/hora durante 24 horas para mantener valores > 100 U/dL, continuando con 2-3 U/kg/hora durante 5-7 días para mantener niveles > 50 U/dL) y para la hemofilia B, concentrado de factor IX recombinante: 120 U/kg, seguido de 50-60 U/kg/cada 12-24 horas, para mantener valores > 40 U/dL, hasta que esté asintomático, continuando con 40-50 U/kg a días alternos durante un total de 10-14 días.
Trombocitopenia neonatal
Los megacariocitos neonatales son más pequeños y con menos núcleos que en los adultos, por lo que la producción plaquetaria fetal-neonatal depende de una proliferación muy activa de los progenitores megacariocíticos, lo que a su vez depende de la actividad de la trombopoyetina (TPO). El ritmo de producción y el turnover son similares al de niños mayores y adultos, y su vida media es de 7-10 días. Las plaquetas neonatales son menos sensibles a factores proagregantes que las de los adultos; el tiempo de hemorragia, sin embargo, es normal, debido a una mayor adherencia y a un hematocrito aumentado. El recuento plaquetario aumenta con la edad gestacional pero, en cualquier caso, debe estar entre 150.000 y 450.000 plaquetas/mm³, aunque un 29% de < 1.500 g al nacer presentan recuento entre 100.000 y 150.000/mm³.
La definición de trombocitopenia en el neonato es la misma que en otras edades: recuento plaquetario inferior a 150.000 plaquetas/mm³ si bien, dado que un 15% de los neonatos sanos tendrán un número de plaquetas entre 100.000 y 150.000 plaquetas/mm³, la mayoría de autores consideran que existe trombocitopenia cuando la cifra es inferior a 100.000 plaquetas/mm³. La trombocitopenia es grave si el número desciende por debajo de 50.000 plaquetas/mm³. En los primeros dos días de vida, alrededor del 1% de los RN tienen menos de 150.000 plaquetas/mm³, un 0,7% tienen menos de 100.000 plaquetas/mm³ y 0,14%, menos de 50.000 plaquetas/mm³. La trombocitopenia es el trastorno hemostático más frecuente en las unidades de cuidados intensivos neonatales, presentándola el 35-50% de los neonatos enfermos (desciende el número de plaquetas entre el 2º y el 4º días de vida, y se incrementa hacia el 10º día) y puede estar causada por una menor producción de plaquetas o por su mayor destrucción o secuestro por acumulación en el interior de un hemangioma, o por una combinación de estos factores. En los RN trombocitopénicos se presentan concentraciones más bajas de trombopoyetina que las observadas en niños mayores con trombocitopenia.
Para la elección del tratamiento tiene importancia la identificación del mecanismo causal de la trombocitopenia. En términos generales, hay mayor probabilidad de que la trombocitopenia aislada sea producida por un mayor consumo de plaquetas que por su menor producción o por secuestro, ya que en estos dos últimos trastornos hay afectación de otras líneas celulares. Es útil también el estudio de un frotis de sangre periférica (cuando es por excesiva destrucción habrá una mayor proporción de plaquetas grandes y jóvenes) y de la médula ósea, para la distinción de estos cuadros. El incremento del tamaño de las plaquetas (mayor de 10,8 fl) y el descenso paralelo en el número de plaquetas sugerirá que hay un consumo excesivo de plaquetas. En un 60% de los casos resulta imposible determinar la causa de la trombocitopenia.
Trombocitopenia inmunitaria
Es casi siempre consecuencia de un trastorno de tipo inmunitario en la madre y el RN tiene un aspecto saludable. Se da en el 0,3% de los RN.
Trombocitopenia neonatal autoinmunitaria
Se debe a la producción de autoanticuerpos IgG contra las plaquetas (púrpura trombocitopénica idiopática, lupus eritematoso sistémico, trastornos linfoproliferativos e hipertiroidismo), que también pasan al feto, ocasionando una disminución del número de plaquetas tanto en el neonato como en su madre. En un 13 a 56% de madres con púrpura trombocitopénica idiopática sus hijos presentan trombocitopenia neonatal, alcanzando las cifras más bajas de plaquetas al quinto día. En un 7-8% de los embarazos se produce una trombocitopenia gestacional benigna, lo que dificulta el diagnóstico, pero raramente ocasiona trombocitopenia neonatal.
Trombocitopenia neonatal aloinmunitaria
Está producida por aloanticuerpos, tras la exposición materna a antígenos plaquetarios ausentes en la madre, heredados del padre, siendo debida a incompatibilidad plaquetaria madre-hijo. Es el equivalente plaquetario de la enfermedad Rh.
Estos antígenos plaquetarios se denominan antígenos plaquetarios humanos (HPA), y las diferentes formas alélicas se distinguen por medio de un sufijo “a” (más frecuente) o “b” (más raro). Generalmente el sistema antigénico HPA-1, contra el antígeno plaquetario HPA-1a (antes denominado P1A1 o Zwa) y el HPA-1b (P1A2 o Zwb), explican el 80-90% de los casos. También se han descrito casos mediados por anticuerpos contra HPA-5 (antígeno HPA-5a o Brb, HPA-5b o Bra), HPA-3 (antígeno HPA3a o Bakb y HPA-3b o Baka) y, más rara vez, HPA-6 (antígeno HPA-6b o Ca). En sujetos asiáticos es frecuente el sistema HPA-4 (antígenos HPA4a o Pena y HPA-4b o Penb). Tiene una incidencia de 1/1.800 RN (1/1.0005.000 RN), produciendo la madre (sobre todo las que poseen el aloantígeno HLA DRW52a, DRB3*0101 o DQB1*0201) anticuerpos contra las plaquetas del hijo, lo que puede acontecer ya en el primogénito. Esto originará trombocitopenia exclusivamente en el niño, tendiendo a normalizarse en 2-3 semanas, pero, entre tanto, un 15-20% presentarán complicaciones, siendo la principal la hemorragia intracraneal. Hay que tener en cuenta que el próximo hijo afecto tendrá un cuadro clínico, como mínimo, tan grave como el del primero, con riesgo de hemorragia intracraneal prenatal especialmente si el anterior hijo la tuvo.
En todos estos cuadros las inmunoglobulinas de la clase IgG no son producidas por el neonato, pero pasan a su circulación desde la madre. Al no ser el anticuerpo autólogo, persistirá la trombocitopenia sólo durante algunos meses, mientras el anticuerpo IgG de la madre permanezca en la circulación del neonato. La vida media de la IgG es de 21 días pero, dado que el anticuerpo se liga a las plaquetas, su vida media depende de las plaquetas sensibilizadas, razón por la cual puede durar solamente horas.
La trombocitopenia puede estar presente al nacer, si bien la caída de las cifras de plaquetas será más acusada entre el segundo y cuarto días de vida. Deben determinarse los aloanticuerpos antiplaquetarios en la sangre materna y, si es posible, el ADN materno y el paterno para determinar los genotipos de los antígenos plaquetarios.
La presentación clínica de la trombocitopenia aloinmune incluye: petequias, hemorragias digestivas, hematuria o hemoptisis y, más raramente, aparece una hemorragia intracraneal que puede originarse prenatal (20-50% de los embarazos afectados) o postnatalmente, y dejar secuelas como hidrocefalia, poroencefalia quística y epilepsia. Los RN con una trombocitopenia autoinmune suelen ser nacidos a término, aparentemente sanos, sin manifestaciones clínicas o, a lo sumo, petequias.
Hay que pensar en esta entidad clínica ante un RN con recuento < 50.000 plaquetas/mm³ en el primer día de vida.
Trombocitopenia inducida por fármacos
Quinina, tolbutamida, hidralazina, metildopa y los diuréticos tiacídicos, administrados a embarazadas, forman anticuerpos de la clase IgG contra un complejo fármaco-hapteno, que muestra reacción cruzada con un antígeno plaquetario, lo que ocasiona trombocitopenia tanto en la madre como en el niño. Otros fármacos administrados a neonatos, especialmente en los pretérmino, se han asociado con trombocitopenia, tales como la indometacina, heparina y vancomicina.
Trombocitopenias no inmunitarias
Incluye a su vez tres subgrupos: Trombopenia infecciosa, Trombopenias no infecciosas, y Trombopenia neonatal de origen genético.
Trombopenia infecciosa
Es debida más bien a una mayor destrucción de plaquetas. Aproximadamente el 60-80% de las sepsis bacterianas cursan con trombocitopenia y un 25%, de forma precoz. Las sepsis por Candida presentan trombopenia en casi el 100% de los casos. Ciertas infecciones víricas también cursan con trombocitopenia (VIH, CMV, parvovirus B19, adenovirus, enterovirus) por una mezcla de producción disminuida y destrucción acelerada. La enterocolitis necrotizante presenta trombopenia en el 80-90% de los casos.
Trombopenias no infecciosas
En este grupo está incluida la trombocitopenia debida a la hipoxia crónica intrauterina que se puede observar en situaciones de insuficiencia placentaria, como en la hipertensión gravídica (1-5% de casos, más frecuente en pretérminos), la diabetes y, a su vez, en alteraciones fetales, como el retraso de crecimiento intrauterino (RCIU) y alteraciones hematológicas (CID, hemangioma gigante o trombosis, por destrucción plaquetar debida a la síntesis de trombina). En estos casos la trombocitopenia es moderada con cifras alrededor de las 50.000-100.000 plaquetas/mm³ y, en el caso del RCIU, las plaquetas se suelen recuperar entre el 7º y 10º día de vida. Otros cuadros o procedimientos que también pueden provocar trombopenia son la anemia hemolítica, exanguinotransfusión, fototerapia y policitemia.
Trombopenia neonatal de origen genético
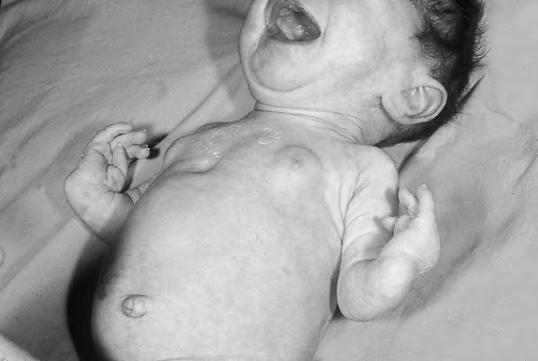
Debida a una mala respuesta a la TPO, asociada habitualmente a síndromes polimalformativos, como en el caso de la trombocitopenia-ausencia de radio (Fig. 2.19.1); o bien a una ausencia de megacariocitos: trombopenia amegacariocítica congénita (TAMCC). La fisiopatología es la disminución de la producción de plaquetas. El inicio es tardío excepto en el caso de TAMCC, que afecta a la época fetal.
El tratamiento de las trombocitopenias neonatales incluye:
En la trombocitopenia aloinmunitaria se aconseja realizar ecografías de control a partir de las 20 semanas de gestación y cordocentesis a los fetos de riesgo, para confirmar la presencia de trombocitopenia y de anticuerpos antiplaquetarios, así como una vía de acceso para administrar intraútero, en caso necesario, plaquetas compatibles, semanalmente o bien inmediatamente antes del parto. Una pauta alternativa es la administración a la madre de inmunoglobulina EV (semanalmente durante el tercer trimestre), con o sin corticoides (prednisona, 20 mg/día, dos semanas antes del parto). Se recomienda la realización de una cesárea electiva si el feto está maduro, pero no existen evidencias de que prevenga la hemorragia intracraneal, por lo que cabe practicar una amniotomía para realizar un recuento plaquetario en sangre del cuero cabelludo fetal, y hacer luego una cesárea sólo cuando dicho recuento sea inferior a 50.000 plaquetas/mm³. Es aconsejable la extracción de 500 mL de sangre completa a la madre inmediatamente antes del parto. Esto permite separar las plaquetas (introduciendo de nuevo los eritrocitos a la madre), lavarlas y suspenderlas de nuevo en plasma tipo AB (60 mL). Si en sangre de cordón hay trombocitopenia, se administrarán las plaquetas de la madre previamente irradiadas. Hay que practicar recuentos sucesivos de plaquetas. Se aconseja iniciar las transfusiones de plaquetas donadas por la madre (10 mL/kg) cuando su número es inferior a 20.000 plaquetas/mm³ si el RN es a término y no tiene factores de riesgo ni hemorragias, pero el umbral para transfundir se eleva a < 50.000 plaquetas/mm³ si es un RN pretérmino, tiene hemorragias o factores de riesgo o presenta hemorragia intracraneal en la ecografía inicial. En las primeras 72-96 horas de vida se deben mantener los recuentos plaquetarios por encima de dichos límites, por ser mayor el riesgo de hemorragia intracraneal durante este periodo.
También puede ser efectiva la administración de gammaglobulina intravenosa (1 g/kg/día, durante 1 a 3 días, o 400 mg/kg/día durante 5 días); si no se produce mejoría se considerará la administración de prednisona o la realización de una exanguinotransfusión con sangre irradiada para retirar los aloanticuerpos maternos.
En la trombocitopenia autoinmune se recomienda que la madre sea tratada acorde a su propio recuento plaquetario, pues no hay predictores válidos para la trombocitopenia grave en el neonato, salvo la determinación de las plaquetas fetales por cordocentesis, pero éste es un procedimiento que tiene riesgos y que no parece justificado en las madres con púrpura trombopénica idiopática (autoanticuerpos contra GPIb/IX) pues sólo aparece la hemorragia intracraneal en un 3% de sus hijos y generalmente es postnatal. Suele aparecer trombocitopenia neonatal cuando la madre tiene < 50.000 plaquetas/mm³. Un 10% de los RN tendrán < 50.000 plaquetas/mm³ y un 4% < 20.000 plaquetas/mm³. Es discutible practicar una amniotomía al término del embarazo para obtener un recuento plaquetario a partir de sangre del cuero cabelludo del feto: si es inferior a 50.000/mm³ está indicada la práctica de cesárea por el mayor riesgo de hemorragia intracraneal en los partos vaginales. Tras el parto es necesario un recuento de plaquetas al neonato dos veces al día; si descienden por debajo de 50.000/mm³ se administrará gammaglobulina EV (1 g/kg); si no se produce mejoría se considerará la administración de prednisona (2 mg/kg/día, dividida en 3 dosis) durante unos pocos días. Si surge una hemorragia grave o las plaquetas descienden por debajo de 30.000 plaquetas/mm³ hay que recurrir a transfusión de plaquetas irradiadas (10-20 mL/kg) y, excepcionalmente, a la exanguinotransfusión.
En la trombocitopenia asociada a hemangiomas gigantes, debida al secuestro y destrucción plaquetaria dentro del tumor vascular, no es necesario tratamiento en ausencia de hemorragias. Si aparecen, deberán administrarse plaquetas y plasma fresco congelado (disminuyen también los niveles de los factores I, V y VIII). Además, debe intentarse disminuir el tamaño del angioma utilizando, en primera instancia, corticoides sistémicos, y si no es suficiente, α2-interferón, cirugía y citostáticos. Se está ensayando el propranolol.
En las trombocitopenias por disminución de la producción, durante los episodios hemorrágicos están indicadas las transfusiones de plaquetas irradiadas (una unidad por cada 5 kg de peso o 10-20 mL, de concentrado de plaquetas/kg). En algunos casos el trasplante de médula ósea ha sido utilizado con éxito.
Perspectivas futuras en el tratamiento. La interleucina 11 es un factor de crecimiento hematopoyético que estimula la proliferación de progenitores de megacariocitos e induce su maduración. En su forma recombinante se ha aprobado para la prevención de la trombocitopenia y la reducción de las transfusiones de plaquetas tras quimioterapia en enfermedades malignas no mieloides y, como efecto adverso se ha referido retención de líquidos. La trombopoyetina recombinante humana (rhTPO) es más eficaz que la interleucina 11 y no cabe esperar un efecto aditivo, pero no aumenta el recuento de plaquetas hasta 3 a 6 días de iniciado el tratamiento, por lo que sólo se beneficiarían los que requieren transfusiones múltiples de plaquetas durante un periodo de varios días y los casos con insuficiente producción de plaquetas.
Hemorragia neonatal por disfunción plaquetaria
Hay que sospechar una disfunción plaquetaria en todo neonato con hemorragias en el que el recuento plaquetario y los estudios de coagulación sean normales. Son más frecuentes los trastornos adquiridos. En general están relacionados con la ingesta materna de fármacos: AAS (la acetilación de la membrana plaquetaria por la aspirina inhibe la liberación de ADP y, por tanto, su agregación), prometacina y promacina. Existen escasos datos concluyentes respecto a los fármacos que producen disfunción plaquetaria al ser administrados a los neonatos, lo mismo que acerca de los factores metabólicos, que pueden influir sobre la función plaquetaria (fototerapia, acidosis, indometacina, penicilina, déficit de ácidos grasos esenciales). Entre los trastornos hereditarios destaca la tromboastenia de Glanzmann. La enfermedad de von Willebrand en su forma clásica tiene un carácter autosómico dominante; cursa con disminución tanto de la actividad coagulante del factor VIII como del antígeno del factor VIII por lo que existirá, además del alargamiento del tiempo de sangría, un tiempo de TPT alargado, siendo tratada como la hemofilia.
Hemorragia asociada con enfermedad hepática
Sus características son similares a las de la CID. El tiempo de protrombina y el TPT están prolongados y no se modifican significativamente tras la administración de vitamina K. La única diferencia respecto a la CID es la existencia en las hepatopatías de un nivel normal del factor VIII. Las principales causas de la disfunción hepática en el periodo neonatal son: citomegalovirus, hepatitis, sífilis, hipoxia, choque, hidrops fetal y nutrición parenteral.
Clínica de las hemorragias neonatales
Hemorragias umbilicales
Son relativamente frecuentes e importantes. Descartada la inadecuada ligadura del cordón se pensará en: déficit de los factores vitamina K dependientes, déficit del factor XIII (es más tardía), hemofilia congénita, alteración del fibrinógeno y patología plaquetaria.
Gastrointestinales
Se manifiestan por hematemesis y melenas. Frecuentemente están motivadas por la ingestión de secreciones hemáticas del canal del parto o por succión de sangre procedente de una grieta del pezón. Estas posibilidades se deben descartar mediante la prueba de Apt, que consiste en mezclar un volumen de heces o del contenido del vómito hemático con cinco volúmenes de agua, centrifugándose la mezcla; se separa el sobrenadante rosa claro (hemolizado) y se agrega 1 mL de hidróxido de sodio al 1% a 4 mL del sobrenadante, observando si existe cambio de color al cabo de dos minutos ya que, mientras la hemoglobina A cambia de color de rosado a pardo-amarillento (lo que indicaría la existencia de sangre materna), la hemoglobina F resiste la desnaturalización y permanece rosada (esto indica que se trata de sangre fetal). La hematemesis aislada neonatal es infrecuente. Se suele deber a deglución de sangre materna o tras epistaxis. Las melenas, de variable intensidad, aparecen generalmente a las 48-72 horas cuando son debidas a déficit de vitamina K, pudiendo ocasionar colapso en casos graves. Existen otras posibilidades de confusión, como la existencia de una mancha roja en el pañal: puede ser debida a la eliminación de ácido úrico por la orina (infarto úrico), sangre procedente de genitales, sobre todo en las niñas por menstruación secundaria al paso placentario de estrógenos maternos o hematuria por ulceración del meato urinario en niños. Son infrecuentes ciertos procesos intestinales causantes de melena, como invaginación intestinal y ulceración gastroduodenal. En ellos serán normales las pruebas de coagulación y el diagnóstico por la imagen aclara las dudas.
Nasales
Aparecen como epistaxis posterior y se manifiestan en forma de hematemesis y melenas.
Respiratorias
No suelen manifestarse como hemoptisis abundantes, sino como espuma sanguinolenta. Aparecen sobre todo en las cardiopatías congénitas, en la asfixia y cuando se practican maniobras bruscas de respiración artificial. Caso especial de este tipo de hemorragias es la hemorragia masiva pulmonar, estudiada en el capítulo anterior.
Genitales
Son frecuentes, en especial durante la circuncisión. La hemorragia vaginal en niñas suele carecer de significación patológica.
Urinarias
En un análisis de orina se encuentra a menudo microhematuria en el RN, pero son raras las macrohematurias. En este caso pueden acompañar al grave infarto renal y a la trombosis venosa renal (riñón grande hematúrico).
Subconjuntivales
Son frecuentes en forma de anillo hemorrágico que rodea la córnea. Son debidas a fragilidad capilar y su pronóstico es bueno.
Cutáneas
En la aparición de petequias o equimosis interviene sobre todo el factor traumático, como sucede en los partos con presentación de nalgas, procidencia de un brazo, cara. También son frecuentes en las trombocitopenias neonatales, incluida la coagulopatía de consumo.
Cefalohematoma y hematoma del esternocleidomastoideo
Son estudiados en un capítulo posterior.
Hemorragias intracraneales
Se describen en un capítulo anterior.
Hemorragia de las suprarrenales
Existen dos formas de presentación: unilateral y bilateral. La primera en la mayoría de las ocasiones es asintomática o sólo cursa con ictericia y se descubre como un hallazgo radiográfico al visualizarse calcificada. La bilateral puede revestir extrema gravedad y se acompaña de síntomas inespecíficos, como hipertermia, cianosis y taquipnea (pseudoneumonía del RN según Golzieher). A veces se podrá palpar como una tumoración abdominal dolorosa, que plantea el diagnóstico diferencial con la trombosis venosa renal y en la urografía en fase inicial se puede demostrar un descenso renal. El diagnóstico se realiza con la ecografía. Evoluciona a menudo hacia la muerte, aunque cabe la regresión con calcificación grosera a las 3-4 semanas, a diferencia de la enfermedad de Wolman (xantomatosis) donde son bilaterales y puntiformes. Generalmente aparece en partos muy traumáticos.
Hemorragias hepáticas
Aparecen también en partos muy distócicos. Suelen estar localizadas por debajo de la cápsula de Glisson, cuya rotura motiva el paso a la cavidad peritoneal, con signos de choque, anemia aguda y hemoperitoneo (curso clínico en dos tiempos).
Hemorragias peritoneales
Son graves, produciendo un cuadro de anemia aguda de difícil diagnóstico. Suelen ser consecutivas a hemorragias hepáticas, suprarrenales, esplénicas o de otros órganos, tras graves distocias. La punción abdominal y la ultrasonografía facilitan el diagnóstico.
Hemorragias de localización pleural
Surgen en neonatos sometidos a maniobras muy traumáticas. El hemoneumotórax es excepcional, pero siempre muy grave.
Hemorragias retinianas
Son un frecuente hallazgo en las exploraciones sistemáticas del fondo de ojo de los RN (34%). Carecen de trascendencia clínica, desapareciendo las pequeñas al cabo de 48 horas.
Las mayores persisten hasta 10 días.
Enfoque diagnóstico del recién nacido con hemorragia
El diagnóstico y tratamiento correctos del RN hemorrágico requieren una síntesis adecuada de los datos obtenidos en los antecedentes familiares y maternos, el examen clínico y unas pruebas de laboratorio cuidadosamente seleccionadas. En un neonato con buen estado general las posibilidades diagnósticas más frecuentes son: deficiencia de vitamina K, trastornos hereditarios de la coagulación, trombocitopenia inmunitaria o inducida por fármacos y traumatismos. En el niño con mal estado general las posibilidades diagnósticas más probables son: CID, enfermedad hepática y consumo mecánico de las plaquetas. Debe correlacionarse el sexo del RN con la información disponible sobre la genética de los trastornos hereditarios de la coagulación: recesiva ligada al sexo (déficit de los factores VIII y IX), autosómica dominante (déficit del factor XI y la enfermedad de von Willebrand), autosómica recesiva (el resto de los déficit factoriales y la variante de la enfermedad de von Willebrand). Es necesario obtener una relación detallada de los fármacos recibidos por la madre así como de los medicamentos administrados al RN, que pueden causar disfunción plaquetaria. Se deberá interrogar a la madre sobre antecedentes de trombocitopenia, padecimiento de rubéola durante el primer trimestre, sífilis o preeclampsia. Especial atención será prestada al tipo y sitio de sangrado, debiendo considerarse los antecedentes de administración o no de vitamina K, traumatismos al nacer, asfixia e inmadurez. En los RN hemofílicos suele haber con mayor frecuencia hematomas cutáneos y subcutáneos, que petequias o equimosis. Si existe sepsis o hepatoesplenomegalia se sospechará que la hemorragia forma parte de un trastorno generalizado que produce trombocitopenia (CID) o alteración de la síntesis hepática de los factores de la coagulación. Se han observado cefalohematomas y hematomas subgaleales en niños con alteraciones congénitas de coagulación y en el déficit de vitamina K. Es frecuente la hemorragia por el muñón umbilical o de la herida de la circuncisión en los casos de déficit de vitamina K y del factor XIII. Debe buscarse la presencia de hemangiomas (síndrome de Kassabach-Merritt) causantes de coagulopatía de consumo.
El primer paso para la evaluación del laboratorio debe ser el recuento de plaquetas con frotis sanguíneo (para ver el tamaño de las plaquetas y la presencia o ausencia de hematíes fragmentados) y la determinación de los tiempos de protrombina (TP), parcial de tromboplastina (TPT) y trombina (TT). Se administrará vitamina K (1-2 mg por vía intravenosa) mientras se esperan los resultados del laboratorio, si ésta no había sido antes administrada profilácticamente.
Un recuento plaquetario disminuido y asociado con una prolongación de los tiempos de protrombina y TPT hará sospechar una CID. Ésta será confirmada con la detección de un aumento de los productos de degradación de la fibrina, alargamiento del tiempo de trombina, disminución de los niveles de los factores V, VIII o I y el hallazgo en el frotis de fragmentación eritrocitaria. Un volumen plaquetario grande (mayor de 10,8 fl) indicaría un aumento de la producción de plaquetas. Si disminuye el recuento de plaquetas en las primeras 72 horas de vida debe hacerse hincapié en los antecedentes maternos (trombocitopenia, trastornos autoinmunitarios, medicamentos, preeclampsia, diabetes, insuficiencia placentaria) y perinatales (asfixia, corioamnionitis). Si la trombocitopenia es tardía resulta más probable la existencia de sepsis nosocomial, enterocolitis necrotizante, trombosis, fototerapia y medicamentos. El marcado plaquetario con indio-111-oxina es útil para determinar la supervivencia de las plaquetas e identificar el lugar de consumo de plaquetas (síndrome de Kassabach-Merritt, trombos intracardiacos o arteriales).
Un recuento plaquetario normal con tiempos de protrombina y TPT alargados indica como diagnóstico más probable el déficit de vitamina K, debiéndose repetir los estudios de TP y TPT pasadas 4 horas de la administración de la vitamina K. En el caso de que continúen prolongados, las posibilidades diagnósticas serían la deficiencia congénita de los factores I, V o X. Cuando la única anomalía de laboratorio en un neonato hemorrágico es la prolongación del TP se deberá considerar la deficiencia congénita del factor II o el VII. Si la única alteración es el alargamiento del TPT las posibilidades diagnósticas son las deficiencias congénitas de los factores VIII, IX, XI o XII, y la enfermedad de von Willebrand.
En ocasiones se pueden producir hemorragias neonatales sin trombocitopenia, ni alteraciones de los tiempos de protrombina o parcial de tromboplastina. Hay que sospechar entonces la deficiencia congénita del factor XIII o un defecto de la función plaquetaria.
El tiempo de hemorragia valora la interacción de las plaquetas con la pared del vaso. Se debe realizar en la superficie pronal del antebrazo, colocando un manguito de esfigmomanómetro proporcional a la talla en la misma extremidad y ajustarlo a una presión de 20 mmHg para RN < 1 kg, 25 mmHg para pesos de 1-2 kg y a 30 mmHg para pesos de más de 2 kg, absorbiendo la sangre con papel de filtro cada 15 segundos hasta que cese la hemorragia, parando la prueba cuando el tiempo excede de 10 minutos. Con esta técnica un tiempo prolongado (> 1,5 minutos en RN a término) indicaría un trastorno cuantitativo o cualitativo de las plaquetas o defectos microvasculares.
No es raro observar estudios anormales de coagulación en RN, debidos a que las muestras estén contaminadas con heparina (utilizada para lavado de catéteres, añadida a las perfusiones) y, con menos frecuencia, por sobredosificación de heparina.
Trastornos trombóticos neonatales
Es una patología poco frecuente aunque se presenta en el RN con una incidencia superior que en niños mayores o adultos, por muchos factores presentes en esta época de la vida. Existen en la literatura dos grandes registros de trombosis neonatales sintomáticas: el alemán, que informa de una incidencia de 5,1/100.000 RN vivos y el canadiense, que da 2,4/1.000 RN ingresados en UCIN. En el 1% de los RN con catéteres vasculares ocurren síntomas sugerentes de complicaciones trombóticas, pero la incidencia de trombosis asintomáticas relacionadas con catéteres vasculares es del 20-30%.
Trastornos trombóticos genéticos. Son las mutaciones de las proteínas C (prevalencia del 0,023), S (prevalencia del 0,037), antitrombina (prevalencia del 0,019), que provoca un déficit de las mismas, y la resistencia a la proteína C activada (debida a una mutación en el factor V denominada FV Leiden). El déficit homocigótico de las proteínas C y S pueden presentarse como lesiones cerebrales y oftálmicas que aparecen intraútero, púrpura fulminante (trombosis vasculares dérmicas que ocasionan necrosis) a las pocas horas o días del parto y, en raras ocasiones, como trombosis de los grandes vasos. Los déficits de AT-III o HCII son raros en el periodo neonatal, pudiendo ocasionar infarto de miocardio al nacer, trombosis aórtica, crisis convulsivas, trombosis de los senos recto y sagital. La mutación del factor V de Leiden es el factor de riesgo protrombótico genético más frecuente, con una prevalencia del 4-6%, y puede ocasionar trombosis arteriales del SNC. Otras trombofilias, como la mutación G20210A de la protrombina (presente en el 1-2% de los caucásicos), deficiencia de metiltetrahidrofolato reductasa y lipoproteína Lp(a), podrían ser factores de riesgo para trombosis neonatal.
Su tratamiento se efectúa con plasma fresco congelado (10-20 mL/kg, cada 6-12 horas) o concentrados de proteína C (60 U/kg), hasta que las lesiones se resuelvan (generalmente, pasadas unas 6-8 semanas), siendo de utilidad para evaluar el tratamiento la medición de los niveles del dímero D. El tratamiento a largo plazo debe efectuarse con anticoagulantes orales, como la warfarina (no se debe administrar en el periodo neonatal), heparina de bajo peso molecular, concentrados de proteína C y trasplante hepático.
Trastornos trombóticos adquiridos. Suelen aparecer en neonatos enfermos (2,4% de los ingresados en UCIN), siendo los catéteres los responsables del 80% de las trombosis venosas y el 90% de las arteriales.
Las trombosis venosas relacionadas con catéter se encuentran en el 2065% de las autopsias de neonatos con catéter venoso umbilical, pudiendo ocasionar embolismo pulmonar, síndrome de la vena cava superior (tumefacción de las extremidades superiores y cabeza), quilotórax (trombosis de la cava o de la vena subclavia), tumefacción de las extremidades inferiores y parte inferior del cuerpo en las trombosis de la vena cava inferior, disfunciones orgánicas (según la colocación del catéter) y secuelas (hipertensión portal, esplenomegalia, hipertensión, síndrome postflebítico). Su diagnóstico se puede hacer por ultrasonografía Doppler o, mejor aún, por venografía. La trombosis de la vena renal puede aparecer en estados patológicos caracterizados por la reducción del flujo renal, hiperviscosidad (poliglobulia, deshidratación), hiperosmolaridad o hipercoagulabilidad; también puede deberse a extensión de trombosis de la vena cava. Cursa con masa en hipocondrio, hematuria, proteinuria y trombocitopenia. Pueden alterarse las pruebas de coagulación y encontrar elevación de los PDF. Debe descartarse un trastorno congénito pretrombótico. El diagnóstico puede efectuarse con la ecografía y el tratamiento debe hacerse con medidas de soporte, anticoagulación con heparina cuando es unilateral y tratamiento trombolítico cuando es bilateral, consiguiéndose supervivencia en más del 95% de los casos.
Las trombosis arteriales relacionadas con catéter se presentan en el 1% de los niños con catéter en arteria umbilical y, de forma asintomática, con mayor frecuencia. El diagnóstico puede hacerse por ecografía Doppler, pero es más sensible y específica la angiografía con contraste. La sintomatología aguda refleja la localización del catéter: hipertensión arterial, necrosis intestinal. Las trombosis aórticas pueden presentarse como blanqueamiento y riego inadecuado que varían desde afectación de un solo dedo del pie hasta afectar a ambas piernas y los glúteos. Las trombosis de la arteria renal se presentan con hipertensión arterial y anuria.
Las trombosis de arterias periféricas se presentan con extremidad fría, pálida, y puede llegar a la gangrena. Pueden ocurrir también trombosis arteriales y venosas espontáneas en vena cava inferior, vena porta, vena hepática, venas adrenales y vasos arteriales. La presencia de hipertensión sistémica en un neonato está frecuentemente relacionada con trombosis de la arteria renal, incluso en ausencia de catéter.
En los casos de trombosis relacionadas con catéteres, se procederá a la retirada del catéter.
Tratamiento de la trombosis
Antes de iniciar cualquier terapia antitrombótica hay que considerar seriamente las potenciales complicaciones que pueden aparecer, más frecuentemente en prematuros, especialmente la hemorragia intracraneal.
Contraindicaciones absolutas serían cirugía del SNC, sangrado activo y procedimientos invasivos en los siguientes 3 días. Para reducir el riesgo de sangrado se mantendrán las cifras de plaquetas por encima de 50.000 plaquetas/mm³ mediante transfusión y el fibrinógeno > 1 g/L, mediante crioprecipitado.
Tratamiento con heparina en el periodo neonatal
Existe una ausencia de consenso para la profilaxis y tratamiento de las complicaciones tromboembólicas en el periodo neonatal. Diversos estudios refieren que, en relación con la heparina estándar, la de bajo peso molecular tiene una farmacocinética más predecible, menores complicaciones hemorrágicas, se administra por vía subcutánea y es igual de eficaz, por lo que se ha convertido en el anticoagulante de elección en el periodo neonatal. La heparina de bajo peso molecular es un glicosaminoglicano de 2.000 a 8.000 daltons de peso molecular; potencia la inhibición del factor Xa, con poco efecto sobre la inhibición de la trombina por la antitrombina. Se recomienda una dosis inicial de 1,5-2 mg/kg cada 12 horas por vía subcutánea de enoxaparina y ajustar la dosis, especialmente en casos de prematuros, hasta alcanzar el límite terapéutico de 0,5-1 unidades/mL de antifactor Xa (medido a las 4 horas de su administración, se recomienda un control semanalmente). Para su administración profiláctica en infusión continua en catéteres centrales se utilizaría la heparina sódica. La última revisión de la Cochrane del 2008 recomienda 0,5 UI/kg/h. En caso de hemorragia se puede utilizar protamina a una dosis de 1 mg/100 U de heparina de bajo peso molecular, pero sólo la neutraliza parcialmente.
Si hay insuficiencia renal la dosis debe disminuirse en el mismo porcentaje que el descenso del filtrado glomerular.
Terapéutica trombolítica en el periodo neonatal
Los trombolíticos funcionan al aumentar la conversión de plasminógeno en plasmina, que después desdobla por proteólisis la fibrina, fibrinógeno, factor V y factor VIII, lo que produce la rotura del coágulo. Debido a que los RN tienen concentraciones bajas de plasminógeno, los trombos neonatales no se lisan con tanta facilidad en respuesta a los fibrinolíticos, lo que hace que, en ocasiones, además de dosis más altas de fibrinolíticos, se deba añadir plasminógeno exógeno en forma de plasma fresco congelado. Se utiliza en casos de trombosis venosa o arterial grave, cuando no es posible la cirugía, cuando el tratamiento anticoagulante no ha sido efectivo o para desobstruir catéteres que se necesita mantener. En neonatos se han utilizado tres fármacos trombolíticos: estreptoquinasa, uroquinasa y rTPA (activador del plasminógeno tisular recombinante). Los dos primeros tienen mayor riesgo de sangrado, por lo que han sido desplazados por rTPA, siendo la dosis recomendada 01-0,6 mg/kg/hora, individualizándose la dosis, controlando el trombo mediante ecografía Doppler. Se debe vigilar TP, TTPA (si < 1,5 se aumentará la dosis y se repetirá al cabo de 4 horas), plaquetas y fibrinógeno. Tras suspender el tratamiento trombolítico se dejará la heparina a dosis de mantenimiento.